

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Review
Volume 13, Number 4, August 2022, pages 177-184

Arrhythmogenic Right Ventricular Cardiomyopathy: The Role of Genetics in Diagnosis, Management, and Screening
Mihir Odaka, e, Steven Douedib, Anton Mararenkoa, Abbas Alshamic, Islam Elkherpitawya, Hani Douedid, Eran Zacksc, Brett Sealovec
aDepartment of Internal Medicine, Jersey Shore University Medical Center, Neptune, NJ 07753, USA
bDepartment of Cardiology, Deborah Heart and Lung Center, Browns Mills, NJ 08015, USA
cDivision of Cardiology, Department of Medicine, Jersey Shore University Medical Center, Neptune, NJ 07753, USA
dDepartment of Cardiology, Community Medical Center, Toms River, NJ 08753, USA
eCorresponding Author: Mihir Odak, Department of Internal Medicine, Jersey Shore University Medical Center, Neptune, NJ 07753, USA
Manuscript submitted April 30, 2022, accepted July 2, 2022, published online August 15, 2022
Short title: Genetics in Arrhythmogenic Cardiomyopathy
doi: https://doi.org/10.14740/cr1373
- Abstract
- Introduction
- Definition and Epidemiology
- Clinical Presentation
- Diagnosis
- Management
- Role of Genetics in Diagnostics and Screening
- Conclusions
- References
| Abstract | ▴Top |
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a predominantly autosomal dominant genetic condition in which fibrous and fatty tissue infiltrate and replace healthy myocardial tissue. This uncommon yet debilitating condition can cause ventricular arrhythmias, cardiac failure, and sudden cardiac death. Management focuses primarily on prevention of syndrome sequelae in order to prevent morbidity and mortality. Genetic testing and screening in affected families, although utilized clinically, has not yet been incorporated in guidelines due to lack of larger studies and data. We aim herein to identify causative gene mutations, present advancements in diagnosis and management, and describe the role of genetic screening and counseling in patients with ARVC. With the advancement of genetic testing and therapy, diseases such as ARVC may become more accurately diagnosed and more effectively managed, ultimately significantly reducing morbidity and mortality.
Keywords: Arrhythmia; Cardiomyopathy; Arrhythmogenic right ventricular cardiomyopathy; Genetics
| Introduction | ▴Top |
Arrhythmogenic right ventricular dysplasia, now known as arrhythmogenic right ventricular cardiomyopathy (ARVC), is identified as an autosomal dominant genetic condition in about 50% of cases with variable penetrance first described in 1982 [1, 2]. Although rare, autosomal recessive forms such as Carvajal syndrome and Naxos disease have also been described. The autosomal dominant form of ARVC has been found to have a prevalence of about 1 in 5,000 in the general population; however, as many individuals are asymptomatic, the true prevalence is likely greater [1]. Primarily described as a desmosomal disease, mutations in non-desmosomal genes also have been identified, making the diagnosis and management of this condition complex and poorly understood [1, 3]. We aim to identify causative gene mutations, present advancements in diagnosis and management, and describe the role of genetic screening and counseling in patients with ARVC.
| Definition and Epidemiology | ▴Top |
ARVC is a genetic disorder in which fibrous and fatty tissue infiltrate and replace healthy myocardial tissue. Clinical phenotypic presentations include ventricular arrhythmias and compromised cardiac output [4]. The underlying etiology of the condition lies in a genetic abnormality of desmosomes that normally allow myocytes to adhere to one another and to function in synchrony with the rest of the cells in the heart [4, 5]. This abnormality leads to progressive ventricular dilation that may affect the right ventricle (RV) or more commonly may have biventricular involvement. While the disease is typically inherited in an autosomal dominant pattern, an autosomal recessive form exists as well with extracardiac manifestations having been described [6]. The condition has been observed to have a low penetrance and varied expressivity of phenotypes [7, 8].
Desmosomes play a crucial role in the proper functioning of the heart. These junctional complexes are a composition of five distinct proteins, which work in tandem to ensure proper mechanical coupling, while gap junctions ensure conduction of electricity and synchronized myocardial contraction. The underlying genes with mutations have been well characterized. Approximately 16 genes (CTNNA3, DES, DSC2, DSG2, DSP, JUP, LMNA, PKP2, PLN, RYR2, TGFB3, TMEM43, TTN, CDH2, SCN5A, and TP63) with individual mutations have been implicated in playing a role in the pathogenesis of ARVC [9]. The mutations result in impaired protein function, thus resulting in myocardial dysfunction and replacement with scar and fat tissue. Of the 16 genes, most mutations have been reported to be amongst the genes that encode for the desmosomes (DSC2, DSG2, DSP, JUP, and PKP2) (Fig. 1) [8, 10, 11].
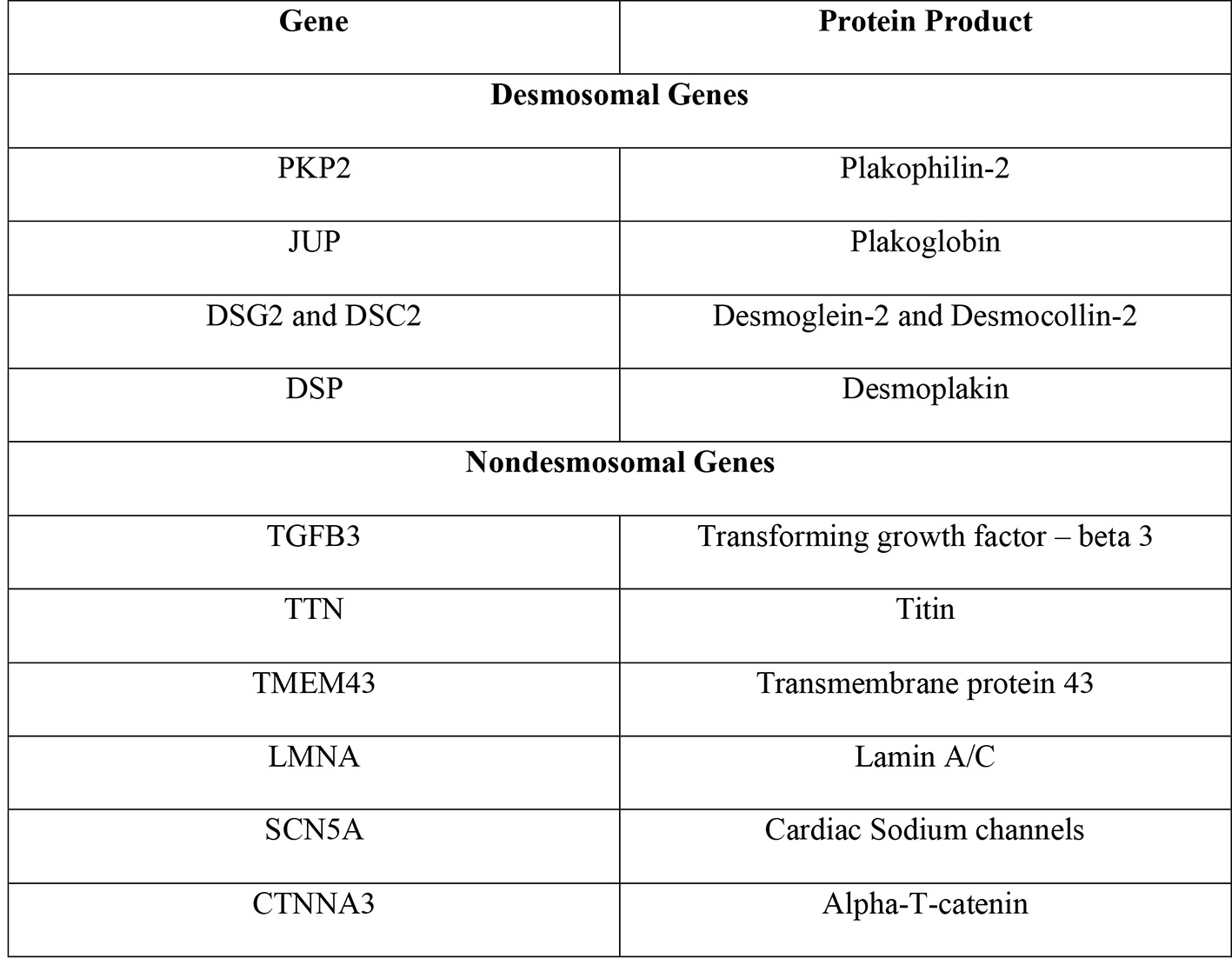 Click for large image | Figure 1. Genes implicated in ARVC and their protein products [8]. ARVC: arrhythmogenic right ventricular cardiomyopathy. |
PKP2 is by far the most frequently involved gene in ARVC. This gene codes for the protein plakophilin-2, which serves as an anchor for the desmoglein proteins in the desmosome complexes that allow cardiac myocytes to adhere to one another (Fig. 2) [8]. A study of 120 individuals found that approximately 26% of individuals were carriers for the PKP2 mutation and among those who had the mutation, this disease expressed low penetrance [8]. A mutation in PKP2 that compromises plakophilin’s structure, however, can lead to instability of desmosomes and dissociation of myocytes from one another. While PKP2 is the most commonly implicated gene in ARVC, an abnormality in any of the desmosomal genes can alter the structure and function of the desmosome and lead to the manifestations of ARVC.
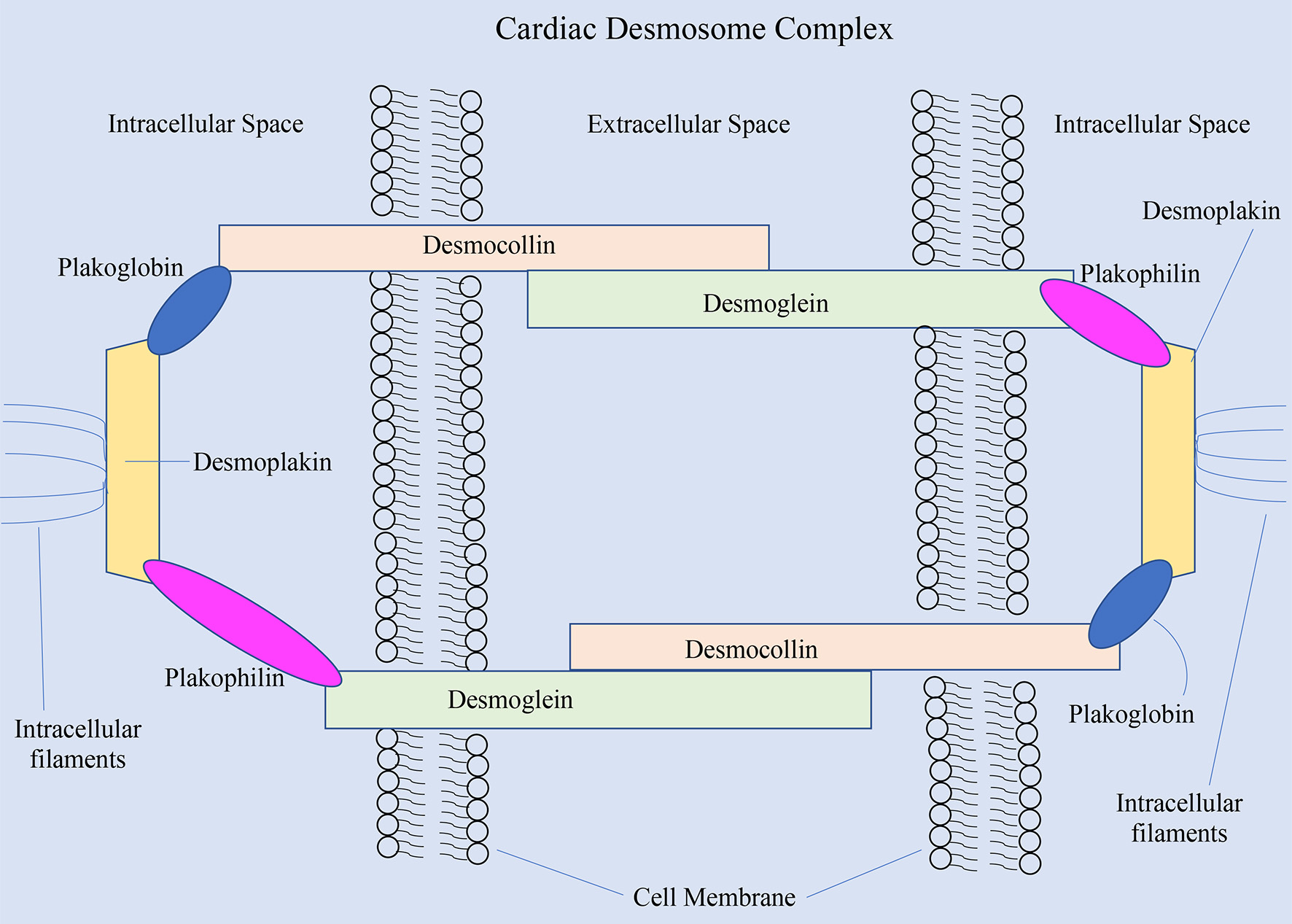 Click for large image | Figure 2. The cardiac desmosome complex and the proteins involved [8]. |
Non-desmosomal genes can contribute to the development of ARVC in several ways. For example, increased expression of transforming growth factor beta 3 (TGFB3) can lead to fibrosis, which was confirmed by a 2005 study of TGFB3 sequencing and expression in those found to have ARVC [8, 12, 13]. Titin mutations also were identified as causative factors in patients and families with ARVC. Transmembrane protein 43 is a protein that works to organize the nuclear membrane in cells; its mutation has been identified as a rare but confirmed cause of ARVC [8]. Additional genes have been found to be involved in the pathogenesis of ARVC with varied mechanisms, including LMNA which serves to stabilize cells and their nuclei, SCN5A which if abnormal can lead to refractory arrhythmias, as well as CTNNA3 which can lead to impaired intercellular adhesion, if altered [8].
In addition to ARVC, a left dominant arrhythmogenic cardiomyopathy (LDAC) also exists, wherein the lateral and postero-lateral aspects of the LV are preferentially involved and early on in the disease course. LDAC can be difficult to differentiate from ARVC, as patients often meet Task Force Criteria for ARVC [14]. ARVC is classically associated with a right bundle branch block compared to left bundle branch block in the case of LDAC. As a result of the ventricular remodeling and loss of myocytes in the LV, an R-wave transition point can be appreciated earlier in the precordial leads in the case of LDAC. The same principles explain the inverted T-wave in the anterolateral leads as the vector of myocardial repolarization is directed towards the RV apex, the side of relative increased muscle mass. The classic epsilon waves seen in ARVC in V1-V3 are instead seen in the inferior/lateral leads in LDAC [15]. As diagnosis of LDAC relies on identifying features of ARVC with concomitant exclusive involvement of the LV, a revision of Task Force Criteria exclusively for LDAC is warranted [16]. ARVC has an approximate prevalence in the United States (US) of 1 in 5,000 individuals [1]. Sudden cardiac death (SCD) is an underreported sequela of ARVC; approximately 20% of SCD cases in individuals younger than 35 years of age are attributed to ARVC [17]. In fact, approximately 10% of SCD cases were found to be due to ARVC based on a series of 86 patients [18]. A study by Bhonsale et al also found SCD to occur in younger individuals with ARVC, and males to have more severe phenotypic expressions [19]. The condition has been studied across numerous populations and has been found to affect a diverse group of people. In the United States specifically, the condition predominantly affects Caucasians, followed by those from Asian or African American backgrounds. Notably, left ventricular involvement was shown to be more prevalent in Italian families with DSP mutations [20]. Recent studies have described non-sustained ventricular tachycardia (VT), left ventricular ejection fraction of < 45%, non-missense mutations, and male sex to be risk factors for malignant arrhythmias. This suggests patients with lamin A/C variant to potentially benefit from a lower threshold for implantable cardioverter-defibrillator (ICD) implantation to prevent lethal arrhythmias [21]. The disease also has been noted to more severely impact males than females. Based on a prospective study of 130 patients who were followed from 1977 to 2000, ARVC resulted in an estimated annual mortality of 2.3% with a total of 24 deaths recorded and with a mean age at death of 54 years (± 19 years). Also noted was that affected individuals with signs of ventricular remodeling and dysfunction and those without arrhythmias carried the worst and best prognoses, respectively [6]. In addition, a study by Groeneweg et al revealed that Task Force Criteria for ARVC were more likely to be met amongst family members with mutations, who are likely to die from cardiogenic causes than those without mutations [22].
An association between ARVC and myocardial remodeling seen in an athlete’s heart has also been described in the literature. It has been proposed that an increased afterload and volume overload may lead to biventricular cardiac dilatation and result in ventricular hypertrophy. Repeated dilatation can result in increased fibroblast activity and lead to focal microvascular deposits of collagen and fibrosis [23, 24]. Another hypothesis raised the concern of an “exercise” induced subtype of ARVC that is underrecognized especially in the younger patient population. The postulation was further revised in 2012 by Trivax et al dubbed the term “Philippides cardiomyopathy” that described the development of myocardial changes that was a result of endurance sport exercises [25]. In their variant, the mechanism was based on the increased myocardial oxygen requirements and resulting increased release of catecholamines that result in free fatty acid metabolites that can lead to diastolic dysfunction, increased production of fibrotic markers, and ultimately result in focal fibrosis [26]. The proposition remains debatable but has been associated with limited data as an independent report noted that one-third of endurance athletes had signs of right ventricular dilatation and hypokinesis [27]. These findings were further supported by Teske and Le Gerche et al that also found disproportionate right ventricular remodeling in endurance athletes [28, 29]. These findings were, however, challenged by Bohm et al in 2016 when they reported no significant difference in RV contractility on the bases of ejection fractions and ventricular volumes amongst endurance athletes [30]. All these reports are limited by small sample sizes, thus stressing the urge for further investigation.
| Clinical Presentation | ▴Top |
ARVC has a large spectrum of presentations and phenotypes, likely due to a large number of genetic mutations that can lead to the syndrome. Regional studies have suggested arrhythmias to generally be the main presenting complaint in ARVC, though this is also postulated to be due to disease progression at the time of diagnosis [31]. Additional presenting complaints reported include palpitations, dyspnea, atypical chest pain, syncope, and lower extremity edema or signs of right heart failure [6, 32]. Symptom onset in the setting of ARVC generally has been hypothesized to be due to activation of the sympathetic nervous system [33]. The remaining patients who receive an ARVC diagnosis are identified as having the syndrome through screening performed in the context of a positive family member, even though they are asymptomatic. Arrhythmias leading to diagnoses of ARVC may originate from the RV, which would yield an electrocardiogram (ECG) tracing signifying VT with a left bundle branch block morphology [33, 34]; however, as several studies have alluded to, this may be an indication of an advanced disease state. These VT episodes are thought to be due to fibrous and fatty infiltration into the myocardium. Studies have also suggested a similar etiology of ARVC as Brugada syndrome, as a sodium channel disorder [35]. This has been suggested by animal models as well, whereby studies have concluded that alterations in cell-cell communicating proteins can lead to altered sodium channel structures and accordingly impact sodium handling in myocardial membranes, leading to arrhythmia [35]. One of the most devastating initial presentations of ARVC can be SCD [34]. Approximately 33% of deaths occurred in patients aged 30 to 39, representing significant mortality and predilection for young people [36]. A robust system to identify these young individuals who are at risk and an adequate screening paradigm remains to be developed to reduce these rates.
Atrial fibrillation is an important comorbidity that is often present amongst patients with ARVC. ARVC is known to induce cardiac remodeling resulting in dilation of the atria. Depending on the specific genetic mutation, alteration of the PKP-2 gene has been proven to impair the sodium currents, thus further contributing to conduction abnormalities. As a result, the risk of atrial arrhythmias (AA) is significantly increased. One report by Chu et al shines light on the prevalence of AA in patients with ARVC [37]. Fortunately, he presented 36 patients - all of which had an ICD thus allowing for continuous monitoring - had shown that the incidence of AA was over 40% with atrial fibrillation accounting for the majority (73%). Atrial fibrillation is a life-threatening comorbidity because it is associated with significantly worse outcomes. The concomitant prevalence of AA and ARVC invariably increases a patient’s risk of developing congestive heart failure, hemodynamically unstable arrhythmias, and potential candidacy for heart transplantation [38]. The prevalence of frequent AA also increases the risk of a patient being inappropriately engaged by an ICD, thus contributing to morbidity [39]. Further investigation by Kikuchi et al provided a retrospective report on 90 patients with ARVC and the incidence of AA as well as heart failure related events over a follow-up period of 29 years. A multivariable analysis had revealed an alarming correlation between worsening decompensated heart failure and patients with AA with underlying ARVC. The rate of hospitalization and mortality was significantly higher amongst patients with underlying AA (hazard ratio: 15.55, 95% confidence interval: 4.82 - 50.17, P < 0.01) [40]. Thus, it is critical for providers to be aware of the potentially life-threatening implications of AA in the setting of ARVC.
| Diagnosis | ▴Top |
Given the clinic presentation and manifestations, ARVC should be considered in young patients with syncope, ventricular arrhythmias, or sudden death [1, 33]. In 1994, the International Task Force proposed a diagnostic criterion for ARVC which was modified by Marcus et al in 2010 [1, 41]. These revised Task Force Diagnostic Criteria utilized 2D echocardiogram, magnetic resonance imaging (MRI), RV angiography, tissue characterization, family history, and arrhythmia/repolarization abnormalities in order to develop major and minor criteria (Fig. 3) [1, 41]. These revised Task Force Criteria include pathologic and biopsy specimen examinations as part of tissue characterization. A definitive diagnosis was defined as satisfying two major, one major and two minor, or four minor criteria in addition to regional wall motion abnormalities present [41].
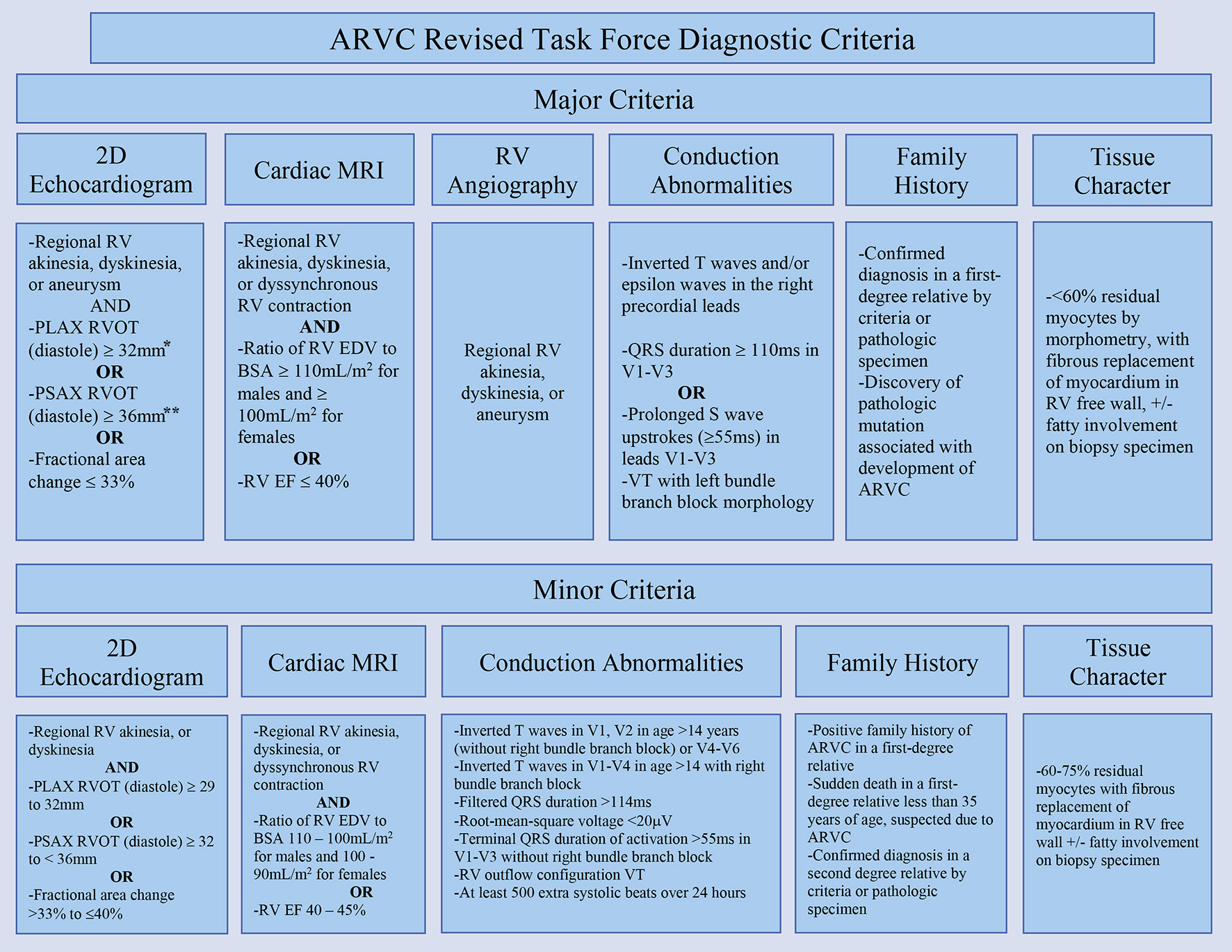 Click for large image | Figure 3. Revised Task Force Criteria for ARVC. *Corrected for body size (PLAX/BSA) ≥ 19 mm/m2 for major criteria and ≥ 16 to ≤ 18 mm/m2 for minor criteria. **Corrected for body size (PSAX/BSA) ≥ 21 mm/m2 for major criteria and ≥ 18 to < 21 mm/m2 for minor criteria [41]. ARVC: arrhythmogenic right ventricular cardiomyopathy; PLAX: parasternal long-axis view; RVOT: right ventricular outflow tract; RV: right ventricle; BSA: body surface area; PSAX: parasternal short-axis view; EDV: end-diastolic volume; EF: ejection fraction. |
Although there may be a high false-negative result, right ventricular endomyocardial biopsy specimens also are used for diagnosis which is described as fibrofatty and/or transmural fibrous replacement of the right ventricular myocardium [41]. In addition, ECG findings such as inverted T-waves and/or epsilon waves (Fig. 4), in the right precordial leads QRS duration of ≥ 110 ms in V1-V3, or prolonged S-wave upstrokes of ≥ 55 ms in leads V1-V3 can be considered major criteria as they are found in up to 90% of diagnosed patients with ARVC [1, 33, 41, 42]. Differential diagnoses such as right ventricular outflow tract tachycardia should be considered, however can be excluded based on the absence of structural heart disease [41]. Electrophysiology studies (EPS) have been found to be of significant benefit in ruling out other differential diagnoses that may cause ventricular arrhythmias [43].
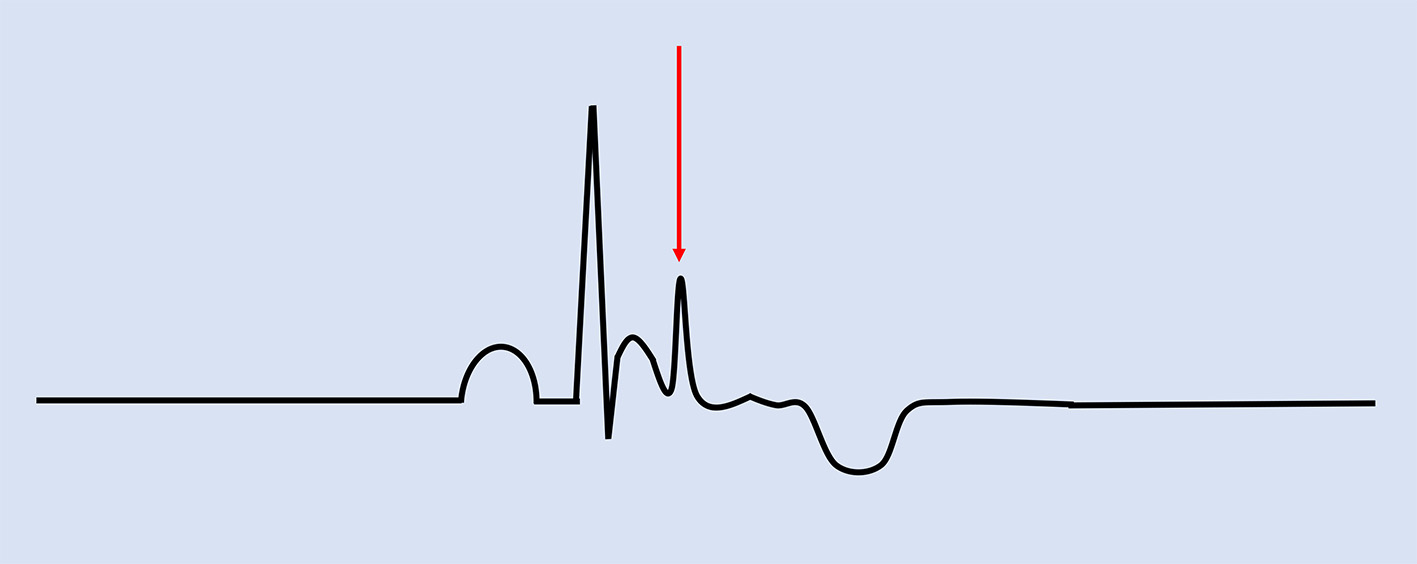 Click for large image | Figure 4. Electrocardiographic findings associated with ARVC. Epsilon waves are characteristic of the condition, indicated by the red arrow. Negative T-waves in V1-V3 are major criteria while prolonged terminal activation duration of greater than 55 ms is a minor criterion for ARVC. ARVC: arrhythmogenic right ventricular cardiomyopathy. |
Genetic testing and screening also have been recommended in individuals with a confirmed first-degree relative who meets the above criteria. In addition, individuals with pathogenic mutations or premature sudden death in a first-degree relative also should be evaluated further [1]. With this in mind, and expanded upon below, genetic screening is fraught with diagnostic challenges due to incomplete penetrance, a wide array of genetic abnormalities, and often no genetic material available on the index relative due to SCD.
| Management | ▴Top |
ARVC is defined as a rare, genetic condition with severe potential complications; however, affected individuals may be asymptomatic. Therefore, management for this syndrome primarily focuses on prevention of associated sequelae. Data on the prevention of SCD with anti-arrhythmic medications, however, are absent and further study is warranted in this area. Antiarrhythmic medications such as beta-blockers and amiodarone have shown some benefit in preventing VT and other life-threatening arrhythmias [4, 5, 43]. Expert consensus on anti-arrhythmic therapy favors the use of beta-blockers, as a class I indication in those with arrhythmia and even a class II indication in individuals even if they do not have arrhythmia. The thought behind this is to suppress adrenergic drive leading to VT in ARVC [41]. While in theory prophylaxis for fatal VT with beta-blockers seems viable, no data on significant mortality benefit have been offered thus far. Expert consensus also endorses the use of amiodarone in conjunction with beta-blocker therapy as a highly effective strategy to reduce the likelihood of precipitating symptomatic VT. While it may alleviate the burden of symptomatic VT, amiodarone’s benefit in preventing SCD is unclear, with research suggesting that treatment with amiodarone or other antiarrhythmic medications alone may not be sufficient to prevent cardiac death, and in such cases may necessitate an ICD [4, 41, 43-45]. Groeneweg et al noted that family members with ARVC mutations were likely to be diagnosed with ARVC based on Task Force Criteria and to die from a cardiac cause. This is supplemented by the fact that SCD was found to be more frequent amongst those without ICDs. Such patients are likely to benefit from beta-blocker and anti-arrhythmic therapy even in the absence of symptoms; however, as Corrado et al noted, a prophylactic ICD may also be reasonable in family members with mutations for ARVC [22]. Ultimately, a comprehensive therapeutic option for ARVC patients who are refractory to other interventions is heart transplantation [1].
| Role of Genetics in Diagnostics and Screening | ▴Top |
The role of genetic screening and testing for ARVC has been difficult to assess. Since genetic mutations are found in about 60% of affected individuals, most experts recommend genetic testing in individuals with first-degree relatives meeting the Task Force Criteria summarized in Figure 2 [1, 46]. In these individuals, due to allelic heterogeneity, sequence analysis has remained the mainstay of genotyping 48]. While reports have concluded comprehensive mutation, screening yields a success rate of about 40%, and costs and time of such analysis have led to barriers in practice [47].
Although there is a lack of established genetic testing options for patients with presumed ARVC, perhaps we can lend from the wealth of knowledge we have on testing for other commonly encountered cardiac conditions. Genetic testing is very well established for several cardiac conditions with a known genetic predisposition such as atrial fibrillation, hypertrophic cardiomyopathy, long QT syndrome and among others.
Cascade screening is being implemented to better identify individuals with a higher risk of developing dyslipidemias. The premise of cascade screening involves testing the first- and second-degree relatives of patients with known dyslipidemia [48]. Although this model resulted in committing individuals to statin therapy from a young age, the small increased risk of experiencing side effects may be counterbalanced by the alarming statistic that less than 10% of high-risk individuals are on optimal, guideline-directed medical therapy [49]. A similar approach may be applied to individuals at high risk for ARVC and thus would qualify them for closer monitoring. A similar approach to cascade screening involves linkage analysis. The benefit of linkage analysis has been well established in conditions such as atrial fibrillation. It is typically performed for individuals with several affected family members. Linkage analysis leverages on the tendency for a known disease-causing gene to be found and, more importantly, to have a high tendency of being inherited with a known genetic marker. This approach has been strategically applied to accurately identify individuals that are known to be at a high risk for atrial fibrillation, long QT syndrome, and hypertrophic cardiomyopathy - conditions that are all known to have specific genetic marker associations [50].
In the case of ARVC, as listed above, we have made significant advances in identifying specific genetic mutations that are known to play a significant role in the pathophysiology of disease progression. Variations in the PKP2 protein are reported to be responsible for ARVC in as many as 30% of cases [51]. The clinical challenge in focusing testing on pure variants stems from the fact that missense mutations are commonly noted and often clinically insignificant in individuals as well. Thus, an abnormality with the protein segment itself does not translate to clinical significance. Larger studies assessing the benefit and optimization of such testing are needed to help identify high-risk individuals at a young age on a cost-effective basis.
| Conclusions | ▴Top |
The role of identification, diagnosis, screening, and management of ARVC has advanced significantly over the last several decades. While anti-arrhythmic medications and ICD placement offer life-saving measures, no curative or genetic therapies have been developed to date. As the medical community continues to advance knowledge of genetic testing and therapy, diseases such as ARVC may be diagnosed more accurately and managed more effectively, ultimately preventing significant morbidity and mortality.
Acknowledgments
None to declare.
Financial Disclosure
The authors declare that they do not have any financial relationship with any commercial entity that has an interest in the subject of this manuscript.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author Contributions
MO, SD, AM, AA, and IE contributed to review of existing literature and writing of the manuscript. HD, EZ, and BS contributed toward project conceptualization.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Cho Y. Arrhythmogenic right ventricular cardiomyopathy. J Arrhythm. 2018;34(4):356-368.
doi pubmed - Orgeron GM, Calkins H. Advances in the diagnosis and management of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Curr Cardiol Rep. 2016;18(6):53.
doi pubmed - Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373(9671):1289-1300.
doi - Gemayel C, Pelliccia A, Thompson PD. Arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2001;38(7):1773-1781.
doi - Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circ Res. 2017;121(7):784-802.
doi pubmed - Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004;110(14):1879-1884.
doi pubmed - Burke AP, Farb A, Tashko G, Virmani R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: are they different diseases? Circulation. 1998;97(16):1571-1580.
doi pubmed - Ohno S. The genetic background of arrhythmogenic right ventricular cardiomyopathy. J Arrhythm. 2016;32(5):398-403.
doi pubmed - Gandjbakhch E, Redheuil A, Pousset F, Charron P, Frank R. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC State-of-the-Art review. J Am Coll Cardiol. 2018;72(7):784-804.
doi pubmed - Quarta G, Muir A, Pantazis A, Syrris P, Gehmlich K, Garcia-Pavia P, Ward D, et al. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation. 2011;123(23):2701-2709.
doi pubmed - Sheikh F, Ross RS, Chen J. Cell-cell connection to cardiac disease. Trends Cardiovasc Med. 2009;19(6):182-190.
doi pubmed - Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36(11):1162-1164.
doi pubmed - Gato A, Martinez ML, Tudela C, Alonso I, Moro JA, Formoso MA, Ferguson MW, et al. TGF-beta(3)-induced chondroitin sulphate proteoglycan mediates palatal shelf adhesion. Dev Biol. 2002;250(2):393-405.
doi pubmed - Imamura Y, Uto K, Nagao M, Nagara K, Yoshizawa S, Kikuchi N, Kimura Y, et al. Characteristics of Left-Dominant Arrhythmogenic Cardiomyopathy. Circ J. 2021;85(12):2245.
doi pubmed - Mascia G, Arbelo E, Porto I, Brugada R, Brugada J. The arrhythmogenic right ventricular cardiomyopathy in comparison to the athletic heart. J Cardiovasc Electrophysiol. 2020;31(7):1836-1843.
doi pubmed - Casella M, Gasperetti A, Sicuso R, Conte E, Catto V, Sommariva E, Bergonti M, et al. Characteristics of patients with arrhythmogenic left ventricular cardiomyopathy: combining genetic and histopathologic findings. Circ Arrhythm Electrophysiol. 2020;13(12):e009005.
doi pubmed - Mu J, Zhang G, Xue D, Xi M, Qi J, Dong H. Sudden cardiac death owing to arrhythmogenic right ventricular cardiomyopathy: Two case reports and systematic literature review. Medicine (Baltimore). 2017;96(47):e8808.
doi pubmed - Zhao YH, Li FH, Jiang HG, et al. Clinicopathological analysis of autopsy of 86 cases with sudden death. Journal of Guodong Medical College. 2009;27:624-628.
- Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JD, Murray B, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015;36(14):847-855.
doi pubmed - Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107(6):700-714.
doi pubmed - Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16(11):e301-e372.
doi pubmed - Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, Murray B, et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet. 2015;8(3):437-446.
doi pubmed - La Gerche A, Claessen G, Dymarkowski S, Voigt JU, De Buck F, Vanhees L, Droogne W, et al. Exercise-induced right ventricular dysfunction is associated with ventricular arrhythmias in endurance athletes. Eur Heart J. 2015;36(30):1998-2010.
doi pubmed - Venlet J, Piers SR, Jongbloed JD, Androulakis AF, Naruse Y, den Uijl DW, Kapel GF, et al. Isolated subepicardial right ventricular outflow tract scar in athletes with ventricular tachycardia. J Am Coll Cardiol. 2017;69(5):497-507.
doi pubmed - Trivax JE, McCullough PA. Phidippides cardiomyopathy: a review and case illustration. Clin Cardiol. 2012;35(2):69-73.
doi pubmed - Trivax JE, Franklin BA, Goldstein JA, Chinnaiyan KM, Gallagher MJ, deJong AT, Colar JM, et al. Acute cardiac effects of marathon running. J Appl Physiol (1985). 2010;108(5):1148-1153.
doi pubmed - Douglas PS, O'Toole ML, Hiller WD, Reichek N. Different effects of prolonged exercise on the right and left ventricles. J Am Coll Cardiol. 1990;15(1):64-69.
doi - Teske AJ, Prakken NH, De Boeck BW, Velthuis BK, Martens EP, Doevendans PA, Cramer MJ. Echocardiographic tissue deformation imaging of right ventricular systolic function in endurance athletes. Eur Heart J. 2009;30(8):969-977.
doi pubmed - La Gerche A, Burns AT, Mooney DJ, Inder WJ, Taylor AJ, Bogaert J, Macisaac AI, et al. Exercise-induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur Heart J. 2012;33(8):998-1006.
doi pubmed - Bohm P, Kindermann W, Meyer T, Scharhag J. Response by Bohm et al to letter regarding article, "Right and left ventricular function and mass in male elite master athletes: a controlled contrast-enhanced cardiovascular magnetic resonance study". Circulation. 2016;134(16):e364-e365.
doi pubmed - Ahmed I, Tipoo FA. Clinical presentation, cardiac magnetic resonance findings, and prognosis of patients with arrhythmogenic right ventricular cardiomyopathy - an experience from Pakistan. J Clin Imaging Sci. 2020;10:48.
doi pubmed - Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376(1):61-72.
doi pubmed - Romero J, Mejia-Lopez E, Manrique C, Lucariello R. Arrhythmogenic right ventricular cardiomyopathy (ARVC/D): a systematic literature review. Clin Med Insights Cardiol. 2013;7:97-114.
doi pubmed - Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112(25):3823-3832.
doi pubmed - Corrado D, Zorzi A, Cerrone M, Rigato I, Mongillo M, Bauce B, Delmar M. Relationship between arrhythmogenic right ventricular cardiomyopathy and Brugada syndrome: new insights from molecular biology and clinical implications. Circ Arrhythm Electrophysiol. 2016;9(4):e003631.
doi pubmed - Tabib A, Loire R, Chalabreysse L, Meyronnet D, Miras A, Malicier D, Thivolet F, et al. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation. 2003;108(24):3000-3005.
doi pubmed - Chu AF, Zado E, Marchlinski FE. Atrial arrhythmias in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and ventricular tachycardia. Am J Cardiol. 2010;106(5):720-722.
doi pubmed - Yeung C, Enriquez A, Suarez-Fuster L, Baranchuk A. Atrial fibrillation in patients with inherited cardiomyopathies. Europace. 2019;21(1):22-32.
doi pubmed - Mazzanti A, Ng K, Faragli A, Maragna R, Chiodaroli E, Orphanou N, Monteforte N, et al. Arrhythmogenic right ventricular cardiomyopathy: clinical course and predictors of arrhythmic risk. J Am Coll Cardiol. 2016;68(23):2540-2550.
doi pubmed - Kikuchi N, Shiga T, Suzuki A, Hagiwara N. Atrial tachyarrhythmias and heart failure events in patients with arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol Heart Vasc. 2020;31:100669.
doi pubmed - Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121(13):1533-1541.
doi pubmed - Zhang L, Liu L, Kowey PR, Fontaine GH. The electrocardiographic manifestations of arrhythmogenic right ventricular dysplasia. Curr Cardiol Rev. 2014;10(3):237-245.
doi pubmed - Corrado D, Wichter T, Link MS, Hauer RN, Marchlinski FE, Anastasakis A, Bauce B, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Circulation. 2015;132(5):441-453.
doi pubmed - Hodgkinson KA, Parfrey PS, Bassett AS, Kupprion C, Drenckhahn J, Norman MW, Thierfelder L, et al. The impact of implantable cardioverter-defibrillator therapy on survival in autosomal-dominant arrhythmogenic right ventricular cardiomyopathy (ARVD5). J Am Coll Cardiol. 2005;45(3):400-408.
doi pubmed - Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, Salerno JU, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108(25):3084-3091.
doi pubmed - Barahona-Dussault C, Benito B, Campuzano O, Iglesias A, Leung TL, Robb L, Talajic M, et al. Role of genetic testing in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Clin Genet. 2010;77(1):37-48.
doi pubmed - Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50(19):1813-1821.
doi pubmed - Singh S, Bittner V. Familial hypercholesterolemia - epidemiology, diagnosis, and screening. Curr Atheroscler Rep. 2015;17(2):482.
doi pubmed - Herman K, Van Heyningen C, Wile D. Cascade screening for familial hypercholesterolaemia and its effectiveness in the prevention of vascular disease. The British Journal of Diabetes & Vascular Disease. 2009;9(4):171-174.
doi - Roselli C, Rienstra M, Ellinor PT. Genetics of atrial fibrillation in 2020: GWAS, genome sequencing, polygenic risk, and beyond. Circ Res. 2020;127(1):21-33.
doi pubmed - Ingles J, Macciocca I, Morales A, Thomson K. Genetic testing in inherited heart diseases. Heart Lung Circ. 2020;29(4):505-511.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.