

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Case Report
Volume 14, Number 5, October 2023, pages 409-415

Associations of MYPN, TTN, SCN5A, MYO6 and ELN Mutations With Arrhythmias and Subsequent Sudden Cardiac Death: A Case Report of an Ecuadorian Individual
Elius Paz-Cruza, f ![]() , Viviana A. Ruiz-Pozoa, f
, Viviana A. Ruiz-Pozoa, f ![]() , Santiago Cadena-Ullauria
, Santiago Cadena-Ullauria ![]() , Patricia Guevara-Ramireza
, Patricia Guevara-Ramireza ![]() , Rafael Tamayo-Trujilloa
, Rafael Tamayo-Trujilloa ![]() , Rita Ibarra-Castillob
, Rita Ibarra-Castillob ![]() , Jose Luis Laso-Bayasb, Paul Onofre-Ruizc, Nieves Domenechd
, Jose Luis Laso-Bayasb, Paul Onofre-Ruizc, Nieves Domenechd ![]() , Adriana Alexandra Ibarra-Rodrigueze, Ana Karina Zambranoa, f, g
, Adriana Alexandra Ibarra-Rodrigueze, Ana Karina Zambranoa, f, g ![]()
aCentro de Investigacion Genetica y Genomica, Facultad de Ciencias de la Salud Eugenio Espejo, Universidad UTE, Quito, Ecuador
bClinical Cardiac Electrophysiologist, Quito, Ecuador
cFacultad de Ciencias de la Salud Eugenio Espejo, Universidad UTE, Quito, Ecuador
dInstituto de Investigacion Biomedica de A Coruna (INIBIC) - CIBERCV, Complexo Hospitalario Universitario de A Coruna (CHUAC), Sergas, Universidad da Coruna (UDC), Spain
eGrupo de Investigacion Identificacion Genetica - IdentiGEN, FCEN, Universidad de Antioquia, Medellin, Colombia
fThese authors contributed equally to this work and share first authorship.
gCorresponding Author: Ana Karina Zambrano, Centro de Investigacion Genetica y Genomica, Facultad de Ciencias de la Salud Eugenio Espejo, Universidad UTE, Quito, Ecuador
Manuscript submitted July 14, 2023, accepted August 2, 2023, published online October 21, 2023
Short title: Arrhythmias and Subsequent SCD
doi: https://doi.org/10.14740/cr1552
| Abstract | ▴Top |
Cardiac pathologies are among the most frequent causes of death worldwide. Regarding cardiovascular deaths, it is estimated that 5 million cases are caused by sudden cardiac death (SCD) annually. The primary cause of SCD is ventricular arrhythmias. Genomic studies have provided pathogenic, likely pathogenic, and variants of uncertain significance that may predispose individuals to cardiac causes of sudden death. In this study, we describe the case of a 43-year-old individual who experienced an episode of aborted SCD. An implantable cardioverter defibrillator was placed to prevent further SCD episodes. The diagnosis was ventricular fibrillation. Genomic analysis revealed some variants in the MYPN (pathogenic), GCKR (likely pathogenic), TTN (variant of uncertain significance), SCN5A (variant of uncertain significance), MYO6 (variant of uncertain significance), and ELN (variant of uncertain significance) genes, which could be associated with SCD episodes. In addition, a protein-protein interaction network was obtained, with proteins related to ventricular arrhythmia and the biological processes involved. Therefore, this study identified genetic variants that may be associated with and trigger SCD in the individual. Moreover, genetic variants of uncertain significance, which have not been reported, could contribute to the genetic basis of the disease.
Keywords: Ventricular arrhythmias; Sudden death; Mutations; NGS
| Introduction | ▴Top |
Sudden cardiac death (SCD) refers to the unexpected death of an individual not attributable to an extracardiac cause. SCD usually happens within the first hour from the onset of symptoms or within 24 h from the last time the individual was seen in good health [1, 2]. It is estimated that approximately 5 million cases of SCD occur worldwide each year [3]. Furthermore, the survival rate is between 27% and 40%, and this percentage has increased due to factors such as cardiopulmonary resuscitation capabilities and external defibrillators. Various causes could trigger SCD, including coronary artery anomalies, structural cardiac anomalies, primary arrhythmias, and coronary artery disease, among others [4].
Cardiac arrhythmias lead to more than one million cases of syncope yearly and are associated with congenital, metabolic, structural, physiological, immunological, and infectious disorders [5, 6]. Among these arrhythmias, ventricular fibrillation (VF) is responsible for the highest number of cases of sudden cardiac arrest (SCA) [7]. VF is a disturbance in the electrical activity of the heart’s lower chambers that results in the absence of an effective heartbeat [8].
Diagnosing arrhythmogenic abnormalities could be complex, as they may manifest as minimal findings in a morphologically normal heart [9]. Moreover, the identification of substrates and triggers of arrhythmias has provided insights into the molecular basis of different pathophysiological pathways [10]. Arrhythmias are diseases with genetic heterogeneity, and as a result, genomic analysis has played a crucial role in the diagnosis of primary arrhythmic disorders by uncovering mutations in different genes correlated with the same disorder [11].
This study describes the case of a man who suffered an aborted SCD, and through genomic analysis, our research group identified variants in genes associated with this cardiac condition.
| Case Report | ▴Top |
Investigations
A 43-year-old Ecuadorian healthy man, with no history of heart disease, suffered an SCD at his workplace 7 years ago. Emergency services resuscitated him, and he was hospitalized. In the hospital, electrocardiogram and echocardiogram were performed, but no structural cardiac cause was found to be associated with the SCD event. The cardiac condition diagnosed was primary VF. Unfortunately, the patient did not respond to medications satisfactorily; thus, an implantable cardioverter defibrillator (ICD) was placed to prevent another sudden death episode. The implanted ICD detected several sudden death events; however, the evolution of the individual’s cardiac condition was favorable. Furthermore, the patient was prescribed an antiarrhythmic drug, amiodarone 200 mg, as a regular medication.
Five years after the SCD incident, the individual was hospitalized due to new syncope events, palpitations, precordial pain, and dyspnea in functional class II/IV. Subsequently, electrocardiogram and echocardiogram were performed, revealing the need for an elective ICD replacement. The discharge diagnoses included New York Heart Association (NYHA) class III heart failure, hemodynamic profile B, and congestive heart failure stage C due to tachycardiomyopathy. A timeline of the relevant episodes of care is depicted in Figure 1.
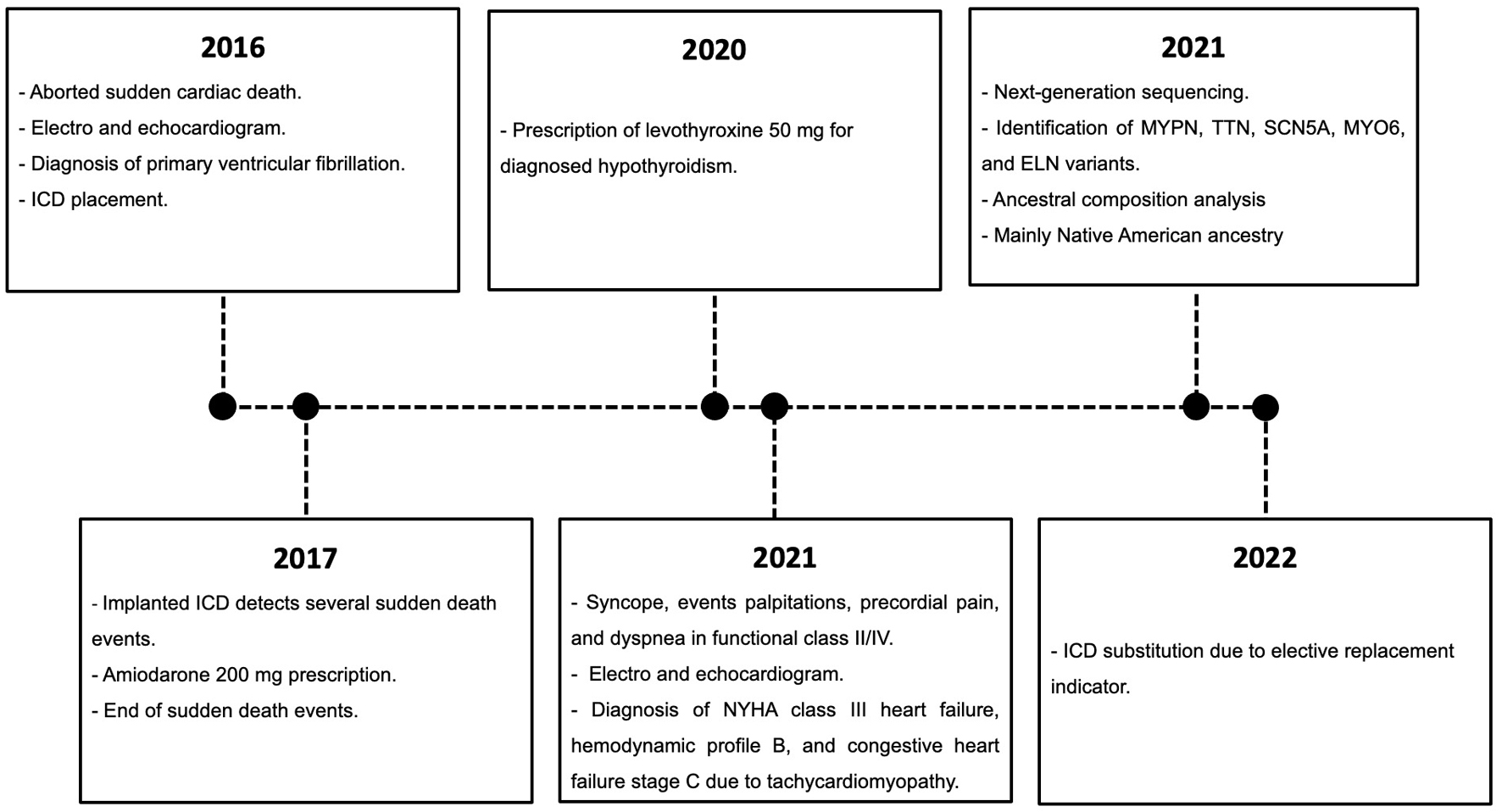 Click for large image | Figure 1. Subject’s episodes of care. The relevant data are displayed in the timeline. |
Diagnosis
Next-generation sequencing (NGS)
A peripheral blood sample was taken, and DNA was extracted using the PureLink™ genomic DNA mini kit. DNA concentrations were quantified using the 1X dsDNA high-sensitivity (HS) and broad range (BR) assay kits on the Qubit™ 4 fluorometer. NGS was performed at the Centro de Investigacion Genetica y Genomica (CIGG) using the TruSight™ cardio (TSC) sequencing panel on the Illumina MiSeq platform. The TSC sequencing panel includes 174 genes with known associations with 17 inherited cardiovascular conditions. For the bioinformatics analyses, DRAGEN Enrichment v3.9.5, Annotation Engine v3.15, PolyPhen, Sift, and Variant Interpreter v2.16.1.300 platforms were used.
Ancestral components determination
Forty-six ancestry-informative INDEL markers (AIMs) were amplified in a multiplex PCR reaction, according to Zambrano et al (2019). Fragment detection was performed on the 3500 genetic analyzer. The results were collected and analyzed on the Data Collection v3.3 and Gene Mapper v.5 platforms. The ancestral analysis was performed using STRUCTURE v.2.3.4 [12].
Outcomes
The coverage was ≥ 50 X on 98.19% of the target regions of the TSC sequencing panel. Variants were classified into five categories (benign, likely benign, variants of uncertain significance (VUS), likely pathogenic, and pathogenic) following the 2015 American College of Medical Genetics and Genomics - Association for Molecular Pathology guidelines [13]. All pathogenic, likely pathogenic, and VUS variants were considered in the analysis (Table 1).
 Click to view | Table 1. Genetic Variants Identified Using TSC Sequencing Panel |
To investigate the cellular processes involving the mutated proteins, protein-protein interaction networks were created using the STRING database [14], with a confidence level of 0.7 (P < 1.0 × 10-16). The network was generated using 34 genes related to the individual’s phenotype and found in the arrhythmia sequencing panel [15] (Supplementary Material 1, www.cardiologyres.org). Notably, the TSC sequencing panel and the arrhythmia sequencing panel share 24 genes. Subsequently, it was determined that of the six genes that presented mutation, five were related to arrhythmias. Among these five genes, only the TTN and SCN5A were present in both sequencing panels.
Furthermore, it has been established that the MYPN protein physically interacts with the TTN protein. Moreover, both the TTN and the SCN5A proteins interact with the CALM1 protein. All these proteins have a physical interaction with other arrhythmia-causing proteins and are involved in the actin filament-based processes (Fig. 2).
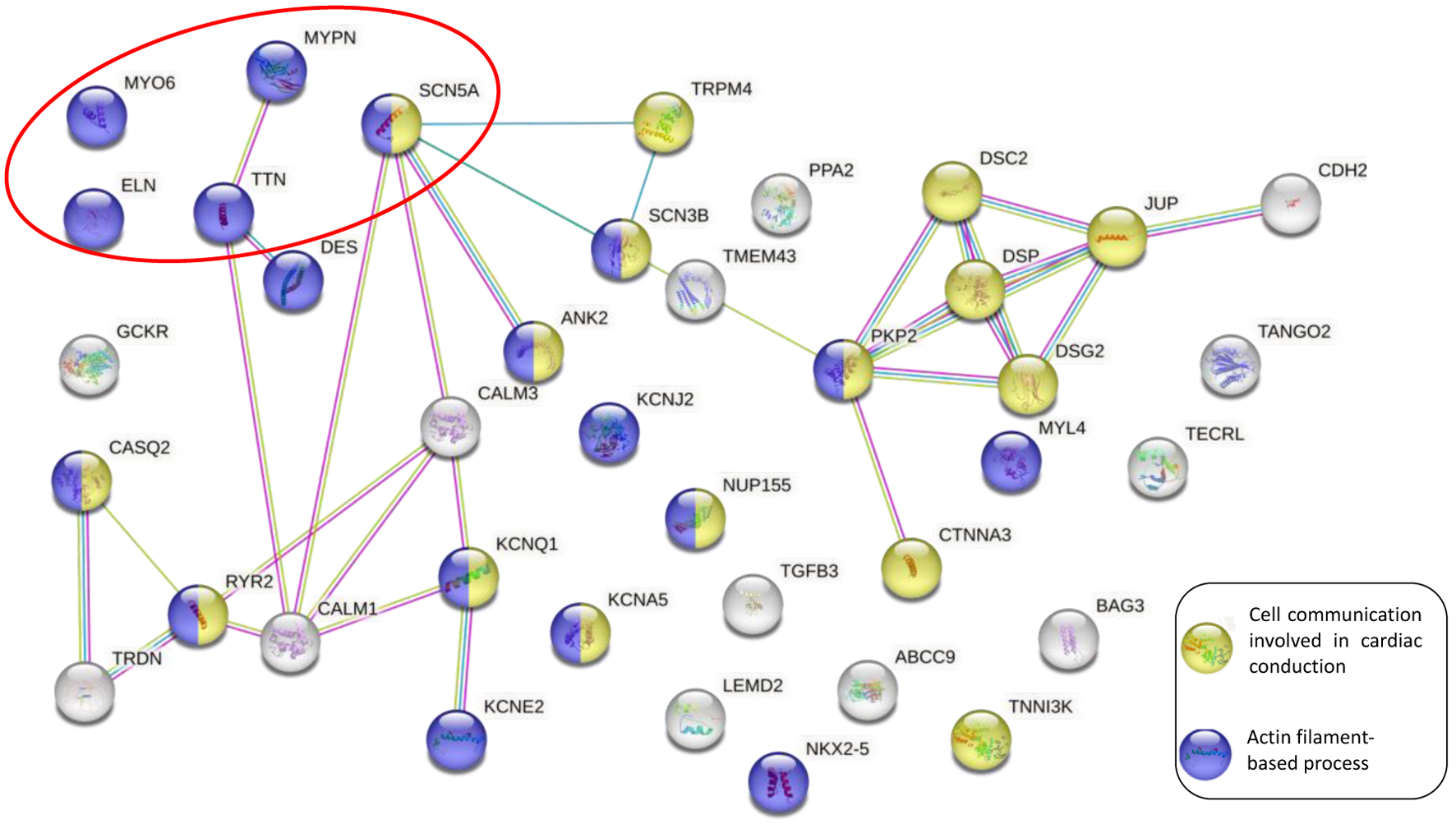 Click for large image | Figure 2. Protein-protein interaction network between the arrhythmia panel genes and the mutated genes on the TSC sequencing panel. The purple circle nodes represent the actin filament-based process. The yellow circle nodes represent cell communication involved in cardiac conduction. The solid lines indicate the physical interaction between proteins, whereas the red circle shows the genes analyzed in the genomic screening. TSC: TruSight™ cardio. |
Moreover, an ancestral composition analysis was performed, and the results showed 2% African, 32.8% European, and 65.2% Native American components.
| Discussion | ▴Top |
In this case report, genomic analyses of a 43-year-old Ecuadorian male with primary VF and subsequent episodes of aborted sudden death were performed. The objective was to determine the association between genetic variants and the individual’s phenotype. Our study identified five mutated genes related to arrhythmias, as well as an unrelated gene. The genomic screening revealed variants in the MYPN, TTN, SCN5A, MYO6, and ELN genes. These variants could show an association between genetic factors, arrhythmias disorders and the increased risk of SCD.
MYPN
Mutations in the sarcomeric myopalladin (MYPN) protein play a significant role in the pathogenesis of cardiac disease. MYPN interacts with several molecules, including α-actinin, located along the stress fibers and in the Z-line of cardiac muscles. Furthermore, the MYPN protein functions as a cytoskeleton support and signaling mediator [16]. Mutations in this protein can affect both contractile (myosin, actin) and non-contractile (titin, Z-disc proteins) proteins of the sarcomere [17].
According to the literature, mutations in the MYPN protein are associated with different types of cardiomyopathies, such as hypertrophic, dilated, and restrictive. In addition, some studies suggest that the MYPN gene network is involved in specific arrhythmia disorders or overlapping phenotypes of inherited cardiomyopathies [18, 19]. However, these types of mutations are rare, accounting for less than 5% [20]. In this genomic screening, a p.(Pro1112Leu) missense MYPN pathogenic variant was described. Although the replacement of the amino acid proline by leucine in this mutation may not be pathogenic, it is essential to consider that the coexistence of other mutations could impact the phenotype [16]. Importantly, the Ecuadorian mestizo individual in this study has not been diagnosed with any of the previously mentioned cardiomyopathies.
SCN5A
The NGS results showed a mutation of uncertain significance at position c.2302A>G (p.Ile768Val) in exon 15 of the SCN5A gene. The SCN5A gene encodes the sodium channel-forming alpha subunit that regulates sodium influx and is involved in the rapid upward depolarization of the action potential. The SCN5A protein, with a molecular weight of 227 kDa, consists of four homologous domains (DI-DIV), and each one is composed of six segments (S1-S6) [21]. Mutations in SCN5A have been associated with various cardiac diseases, such as ventricular arrhythmias, sudden death syndrome, and cardiac conduction disturbances [22].
ELN
The human elastin gene, ELN, is part of the extracellular matrix and is involved in the elasticity and strength of tissues such as arterial vessels, lungs, and others [23]. Elasticity allows blood vessels to perform the process of diastole and systole for optimal heart function. In addition, in arteries, elasticity maintains tone and regulates blood pressure [24].
Mutations in the ELN gene are associated with diseases such as supravalvar aortic stenosis (SVAS), characterized by significant narrowing of the large arteries [25, 26]. In the genomic analysis performed in the individual, a mutation of uncertain significance was identified at position c.2142_2156 of (p.Gly715_Val719del), resulting in an in-frame deletion in exon 30 of the ELN gene. Mutations in the ELN gene may be associated with a sporadic or inherited autosomal dominant SVAS.
In 2016, Latham et al mentioned that the SCD risk in patients with SVAS is increased compared to non-syndromic SVAS patients [27]. Therefore, it is essential to understand this type of mutation in patients diagnosed with SVAS, particularly for medical examinations or surgeries that require sedation, since there have been cases of SCD leading to the fatal outcomes [23].
MYO6
MYO6 gene encodes a protein that moves toward the minus end of actin filaments. The protein has a motor domain with ATP and actin-binding sites and a globular tail that interacts with other proteins. Mutations in the motor domain of myosin could cause cardiac problems, such as dilated and hypertrophic cardiomyopathies [28], as well as different forms of congenital heart defects [29]. However, our patient does not present any of these alterations.
TTN
The TTN gene encodes a large and abundant protein found in striated muscle. This protein is divided into two regions: the N-terminal I-band, which is the elastic part, and the C-terminal A-band that acts as a regulatory protein. The N-terminal region of the Z-disk and the C-terminal region of the M-line bind to the Z-line and the M-line of the sarcomere, respectively. Additionally, TTN contains binding sites for muscle-associated proteins, contributing to the assembly of the contractile machinery in muscle cells [30].
TTN variants are the leading cause of dilated and familial hypertrophic cardiomyopathies and have also been associated with an increased risk of ventricular arrhythmias [31]. However, it is important to highlight that our patient does not present any of these cardiomyopathies.
GCKR
The heterozygous mutation p.(Val103Met) of the glucokinase regulatory gene (GCKR) is another variant found in this study as likely pathogenic. The GCKR gene encodes the glucokinase regulatory protein (GCK), which acts as a glucose regulator in hepatocytes and β cells of the pancreas [32, 33].
According to a kinetic assay characterization of the p.Val103Met variant, it was classified as a severe loss-of-function variant due to its ability to inhibit GCK protein activity [32]. This mutation, p.Val103Met, was found in non-Hispanic individuals of mixed European ancestry, according to the study by Rees et al (2012). While some research describes the association of GCK function with coronary artery disease and ischemic stroke [34], there is no evidence of an association with arrhythmias in the literature.
Protein-protein interaction network
The protein-protein interaction network of physical protein interactions and biological processes can provide valuable information about the relationship with arrhythmia processes. For instance, the actin filament-based biological process, as shown in Figure 2, has been described by Camors et al (2022). The authors mention that changes in actin protein expression are related to the progressive reduction of the right ventricular contraction and development of arrhythmias in a premature phase of arrhythmogenic cardiomyopathy [35].
In some genetic cardiomyopathies, clinical variability is observed even among patients with similar genotypes, ranging from asymptomatic cases to heart failure. This heterogeneity suggests that other factors play a significant role in modifying the clinical phenotype, potentially influencing disease potentiation or protection. These factors include modulatory genes, polymorphism, other unknown genes, as well as environmental and endogenous factors (age, sex, physical exercise, drugs, hormones, viral infections, and emotional stress) [36].
Furthermore, studies suggest that the combination of genetic variants and the additive effect of mutations in different genes could potentially trigger a more aggressive phenotype in the context of cardiac arrhythmias [37].
Conclusion
SCD is often caused by cardiac disorders that can remain asymptomatic and difficult to diagnose. Genomic screening is a valuable tool that could reveal pathogenic, likely pathogenic, and VUS genetic variants related to a phenotype. The genetic variants identified in the Ecuadorian individual, particularly in the MYPN, GCKR, TTN, SCN5A, MYO6, and ELN genes, may be related to cardiac diseases that could be associated with primary VF and SCD. It is also possible that the genetic variants found could have an additive effect on the development of arrhythmia diseases.
Therefore, the identification of these variants could significantly contribute to the comprehension of the genetic basis of arrhythmias and, by extension, SCD. In addition, it would be important to expand the study to encompass other individuals afflicted with the same cardiac condition, to ascertain any potential association with the genetic variants examined in this research article.
| Supplementary Material | ▴Top |
Suppl 1. Genes and phenotypes associated of the arrhythmia and TruSight™ Cardio sequencing panels.
Acknowledgments
We are grateful to the Universidad UTE for supporting the researchers.
Financial Disclosure
The experimentation and publication fee of this article are funded by Universidad UTE.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
The participant provided their written informed consent to participate in this study. The study conducted with human participants followed the ethical standards of the 2013 Helsinki Declaration and was approved by the Committee on Ethics and Research in Human Subjects (CEISH)-UTE University (protocol code CEISH-2021-016, date of approval 18-05-2022).
Author Contributions
Conceptualization: EPC, VARP and AKZ. Methodology: EPC, VARP, AKZ, SCU, PGR, RTT, JLLB, ND, AAIR, and POR. Writing - original draft preparation: EPC and VARP. Writing - review and editing: EPC, VARP and SCU. Supervision: AKZ. Project administration: AKZ. Funding acquisition: AKZ. All authors read and approved the final manuscript.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
AIMs: ancestry-informative INDEL markers; GCKR: glucokinase regulatory gene; HS: high-sensitivity; ICD: implantable cardioverter defibrillator; MYPN: sarcomeric myopalladin; NGS: next-generation sequencing; NYHA: New York Heart Association; SCA: sudden cardiac arrest; SCD: sudden cardiac death; SVAS: supravalvar aortic stenosis; TSC: TruSight™ Cardio; VF: ventricular fibrillation; VUS: variant of uncertain significance; ACS: acute coronary syndrome; BMI: body mass index; CABG: coronary artery bypass grafting; CAD: coronary artery disease; CKD: chronic kidney disease; COPD: chronic obstructive pulmonary disease; ESRD: end-stage renal disease; HIV: human immunodeficiency virus; LAD: left anterior descending; PCI: percutaneous coronary intervention; PE: pulmonary embolism; STEMI: ST-elevation myocardial infarction
| References | ▴Top |
- Markwerth P, Bajanowski T, Tzimas I, Dettmeyer R. Sudden cardiac death-update. Int J Legal Med. 2021;135(2):483-495.
doi pubmed pmc - Kuriachan VP, Sumner GL, Mitchell LB. Sudden cardiac death. Curr Probl Cardiol. 2015;40(4):133-200.
doi pubmed - Chugh SS. Early identification of risk factors for sudden cardiac death. Nat Rev Cardiol. 2010;7(6):318-326.
doi pubmed pmc - Durante A, Laforgia PL, Aurelio A, Foglia-Manzillo G, Bronzato S, Santarone M, Corrado G. Sudden cardiac death in the young: the bogeyman. Cardiol Young. 2015;25(3):408-423.
doi pubmed - Campuzano O, Sanchez-Molero O, Fernandez A, Iglesias A, Brugada R. Sudden cardiac death of arrhythmic origin: Value of post-mortem genetic analysis. Spanish Journal of Legal Medicine. 2018;44(1):32-37.
doi - Vukmir RB. Cardiac arrhythmia diagnosis. Am J Emerg Med. 1995;13(2):204-210.
doi pubmed - Rodriguez-Reyes H, Munoz Gutierrez M, Marquez MF, Pozas Garza G, Asensio Lafuente E, Ortiz Galvan F, Lara Vaca S, et al. [Sudden cardiac death. Risk stratification, prevention and treatment]. Arch Cardiol Mex. 2015;85(4):329-336.
doi pubmed - Aras KK, Kay MW, Efimov IR. Ventricular fibrillation: rotors or foci? Both! Circ Arrhythm Electrophysiol. 2017;10(12):e006011.
doi pubmed pmc - Rodriguez-Calvo MS, Brion M, Allegue C, Concheiro L, Carracedo A. Molecular genetics of sudden cardiac death. Forensic Sci Int. 2008;182(1-3):1-12.
doi pubmed - Spears DA, Gollob MH. Genetics of inherited primary arrhythmia disorders. Appl Clin Genet. 2015;8:215-233.
doi pubmed pmc - Wilde AA, Bezzina CR. Genetics of cardiac arrhythmias. Heart. 2005;91(10):1352-1358.
doi pubmed pmc - Zambrano AK, Gaviria A, Cobos-Navarrete S, Gruezo C, Rodriguez-Pollit C, Armendariz-Castillo I, Garcia-Cardenas JM, et al. The three-hybrid genetic composition of an Ecuadorian population using AIMs-InDels compared with autosomes, mitochondrial DNA and Y chromosome data. Sci Rep. 2019;9(1):9247.
doi pubmed pmc - Laboratories KD, et al. Standards and guidelines for the interpretation of sequence variants. Acta Ophthalmol. 2018;96(S261):134-134.
doi - Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, Gable AL, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638-D646.
doi pubmed pmc - Hiippala A, Tallila J, Myllykangas S, Koskenvuo JW, Alastalo TP. Expanding the phenotype of Timothy syndrome type 2: an adolescent with ventricular fibrillation but normal development. Am J Med Genet A. 2015;167A(3):629-634.
doi pubmed - Mastroianno S, Palumbo P, Castellana S, Leone MP, Massaro R, Potenza DR, Mazza T, et al. Double missense mutations in cardiac myosin-binding protein C and myopalladin genes: A case report with diffuse coronary disease, complete atrioventricular block, and progression to dilated cardiomyopathy. Ann Noninvasive Electrocardiol. 2020;25(3):e12687.
doi pubmed pmc - Refaat MM, Hassanieh S, Ballout JA, Zakka P, Hotait M, Khalil A, Bitar F, et al. Non-familial cardiomyopathies in Lebanon: exome sequencing results for five idiopathic cases. BMC Med Genomics. 2019;12(1):33.
doi pubmed pmc - Gu Q, Mendsaikhan U, Khuchua Z, Jones BC, Lu L, Towbin JA, Xu B, et al. Dissection of Z-disc myopalladin gene network involved in the development of restrictive cardiomyopathy using system genetics approach. World J Cardiol. 2017;9(4):320-331.
doi pubmed pmc - Huby AC, Mendsaikhan U, Takagi K, Martherus R, Wansapura J, Gong N, Osinska H, et al. Disturbance in Z-disk mechanosensitive proteins induced by a persistent mutant myopalladin causes familial restrictive cardiomyopathy. J Am Coll Cardiol. 2014;64(25):2765-2776.
doi pubmed pmc - Herrera-Rodriguez DL, Totomoch-Serra A, Rosas-Madrigal S, Luna-Limon C, Marroquin-Ramirez D, Carnevale A, Rosendo-Gutierrez R, et al. Genes frequently associated with sudden death in primary hypertrophic cardiomyopathy. Arch Cardiol Mex. 2020;90(1):58-68.
doi pubmed - Li W, Yin L, Shen C, Hu K, Ge J, Sun A. SCN5A variants: association with cardiac disorders. Front Physiol. 2018;9:1372.
doi pubmed pmc - Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, George AL, Jr., et al. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117(15):1927-1935.
doi pubmed pmc - Watts CR, Awan SN, Marler JA. An investigation of voice quality in individuals with inherited elastin gene abnormalities. Clin Linguist Phon. 2008;22(3):199-213.
doi pubmed - Osei-Owusu P, Knutsen RH, Kozel BA, Dietrich HH, Blumer KJ, Mecham RP. Altered reactivity of resistance vasculature contributes to hypertension in elastin insufficiency. Am J Physiol Heart Circ Physiol. 2014;306(5):H654-666.
doi pubmed pmc - Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MT. Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet. 1997;6(7):1021-1028.
doi pubmed - Markush D, Sanchez-Lara PA, Grand K, Wong R, Garg R. Sudden cardiac arrest during a sedated cardiac magnetic resonance study in a nonsyndromic child with evolving supravalvar aortic stenosis due to familial ELN mutation. Pediatr Cardiol. 2023;44(4):946-950.
doi pubmed pmc - Latham GJ, Ross FJ, Eisses MJ, Richards MJ, Geiduschek JM, Joffe DC. Perioperative morbidity in children with elastin arteriopathy. Paediatr Anaesth. 2016;26(9):926-935.
doi pubmed - Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez-Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3(2):155-161.
doi pubmed pmc - Posch MG, Waldmuller S, Muller M, Scheffold T, Fournier D, Andrade-Navarro MA, De Geeter B, et al. Cardiac alpha-myosin (MYH6) is the predominant sarcomeric disease gene for familial atrial septal defects. PLoS One. 2011;6(12):e28872.
doi pubmed pmc - Evans WN, Acherman RJ, Law IH, Von Bergen NH, Samson RA, Restrepo H. Neonatal complex arrhythmias possibly related to a TTN mutation. J Neonatal Perinatal Med. 2017;10(3):343-346.
doi pubmed - Enriquez A, Liang J, Smietana J, Muser D, Salazar P, Shah R, Badhwar N, et al. Substrate characterization and outcomes of ventricular tachycardia ablation in TTN (Titin) cardiomyopathy: a multicenter study. Circ Arrhythm Electrophysiol. 2021;14(9):e010006.
doi pubmed - Shetty S, Xing C, Garg A. Type 1 hyperlipoproteinemia due to compound heterozygous rare variants in GCKR. J Clin Endocrinol Metab. 2016;101(11):3884-3887.
doi pubmed pmc - Zhou YJ, Hong SC, Yin RX, Yang Q, Cao XL, Chen WX. Polymorphisms in the GCKR are associated with serum lipid traits, the risk of coronary artery disease and ischemic stroke. Int J Clin Exp Med. 2015;8(7):10678-10686.
pubmed pmc - Shen H, Pollin TI, Damcott CM, McLenithan JC, Mitchell BD, Shuldiner AR. Glucokinase regulatory protein gene polymorphism affects postprandial lipemic response in a dietary intervention study. Hum Genet. 2009;126(4):567-574.
doi pubmed pmc - Camors EM, Roth AH, Alef JR, Sullivan RD, Johnson JN, Purevjav E, Towbin JA. Progressive reduction in right ventricular contractile function attributable to altered actin expression in an aging mouse model of arrhythmogenic cardiomyopathy. Circulation. 2022;145(21):1609-1624.
doi pubmed pmc - Kim KH, Pereira NL. Genetics of cardiomyopathy: clinical and mechanistic implications for heart failure. Korean Circ J. 2021;51(10):797-836.
doi pubmed pmc - Coll M, Perez-Serra A, Mates J, Del Olmo B, Puigmule M, Fernandez-Falgueras A, Iglesias A, et al. Incomplete penetrance and variable expressivity: hallmarks in channelopathies associated with sudden cardiac death. Biology (Basel). 2017;7(1):3.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.