

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website http://www.cardiologyres.org |
Case Report
Volume 7, Number 5, October 2016, pages 178-180
Cardiac Amyloidosis Masked as Hypertrophic Cardiomyopathy: A Case Report
Jing Wanga, b, Amy Marzolfa, Janet C. L. Zhanga, Anjali Owensa, Yuchi Hana, c
aCardiovascular Division, Department of Medicine, Perelman School of Medicine of the University of Pennsylvania, Philadelphia, PA, USA
bDepartment of Cardiology, PLA General Hospital, Beijing, China
cCorresponding Author: Yuchi Han, Cardiovascular Division, Hospital of the University of Pennsylvania, Room 9022 East Gates, 3400 Spruce Street, Philadelphia, PA 19104-4283, USA
Manuscript accepted for publication September 30, 2016
Short title: Cardiac Amyloid
doi: https://doi.org/10.14740/cr496w
| Abstract | ▴Top |
It is well known that cardiac amyloidosis and hypertrophic cardiomyopathy (HCM) have different physiologies and pathologies. However, it might be difficult to differentiate cardiac amyloidosis from HCM in certain clinical situations.
Keywords: Cardiac amyloid; Hypertrophic cardiomyopathy
| Introduction | ▴Top |
Both hypertrophic cardiomyopathy (HCM) and amyloidosis present with increased myocardial thickness and mass. The delayed enhancement patterns on cardiac magnetic resonance imaging (MRI) are thought to distinguish these two diseases. However, we report a case where the patient had patchy delayed enhancement and was thought to have HCM but then found to have amyloid pattern 5 months later. The lesson to be learned here is that in early stages of cardiac amyloid, a patient could present with less apparent delayed enhancement patterns and the imaging findings have to be correlated with the patient’s clinical presentation in order to make a correct diagnosis.
| Case Report | ▴Top |
A 64-year-old female presented with recurrent chest pain and progressive shortness of breath in April 2015. She had a history of asthma and panic attacks. The worsened symptoms triggered a cardiac workup which included cardiac catheterization which showed no obstructive lesions. The findings on echocardiography and cardiac magnetic resonance (CMR) in 2015 suggested the diagnosis of HCM (Table 1). The patient did not have a family history of HCM, but her father died suddenly at age 48. Her paternal grandfather and two paternal uncles all died suddenly before the age of 40. Genetic testing was negative for pathogenic variants. A serum protein electrophoresis and urine protein electrophoresis were unrevealing. Her breathlessness had improved with a diuretic regimen of low dose furosemide and aldactone with stable volume status. As part of the follow-up, she was re-examined in February 2016, at which time ECG showed low voltage and echocardiography demonstrated restrictive filling pattern. Repeated CMR provided the most important diagnostic information, which is the pattern of abnormal subendocardial late gadolinium enhancement (LGE) suggesting cardiac amyloidosis (Fig. 1). Based on CMR findings, a fat pad biopsy was completed, but negative. A subsequent endomyocardial biopsy was positive for amyloidosis.
 Click to view | Table 1. Characteristics of Examination Findings |
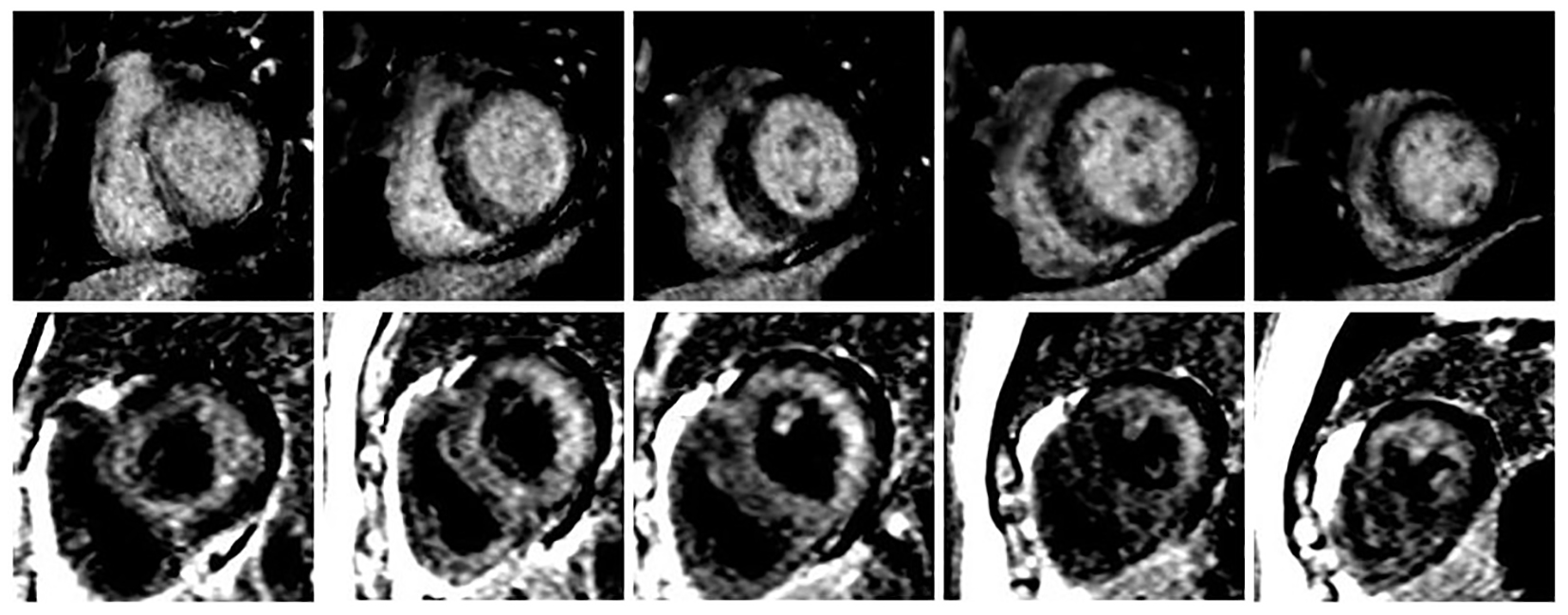 Click for large image | Figure 1. Different LGE patterns between two CMRs. The upper panel showed mid-myocardial delayed enhancement in first CMR (October 2015), and the lower panel showed global subendocardial enhancement in second CMR (March 2016). |
| Discussion | ▴Top |
To the authors’ knowledge, such a dramatic change in CMR LGE pattern from faint patchy mid-myocardial enhancement to global subendocardial enhancement in 6-month follow-up has not been reported. This case raises an important question: could early stage of amyloidosis begin with cardiac hypertrophy without significant amyloid deposition elsewhere and thus could be confused with HCM?
Amyloidosis is a rare protein deposition disease caused by misfolding followed by tissue deposition of highly ordered aggregate proteins forming amyloid fibrils leading to progressive organ failure [1]. The definitive diagnosis should be made by endomyocardial biopsy.
The QRS amplitude on ECG is a useful discriminator in infiltrative versus HCM, since voltages are usually increased in muscular hypertrophy and diminished in the infiltrative myopathies. For our case, the voltage showed a decreasing course between the two examinations. Of note, the QRS voltage was normal on the baseline ECG, not increased, which is atypical for HCM, but certainly does not exclude HCM.
CMR is becoming the standard imaging modality for the diagnosis of cardiac amyloidosis. The goals of a comprehensive CMR evaluation of cardiac amyloidosis should include evaluation of cardiac function and myocardial characterization. The classic CMR findings demonstrate an endocardial-epicardial signal intensity gradient typical for heart amyloidosis, with the early gadolinium clearance from the blood pool [2]. LGE patterns could be divided into no LGE, global subendocardial involvement, and transmural LGE. Transmural LGE was significantly associated with mortality (hazard ratio, 4.1; 95% confidence interval, 1.3 - 13.1; P < 0.05) [3]. Our case showed global subendocardial enhancement on the repeat CMR, which is different from patchy mid-myocardial delayed enhancement in first CMR. Amyloid deposition essentially restructures the heart and restricts it from stretching and properly filling with blood. Our case showed the global hypokinesis and further decreased left ventricular ejection fraction in the repeat CMR, which may have reflected the progression of disease.
The findings on this case suggest that at the early stage, cardiac amyloid may demonstrate hypertrophy as the only imaging feature without significant abnormalities on LGE. It is only after the accumulation of the infiltrative substances for a period of time that the characteristics of cardiac amyloidosis on CMR begin to appear and could be detected. We have the chance to observe the progressive course in this case and place it on the continuum of amyloid accumulation as determined by LGE pattern progressing from normal to transmural [4].
Funding Sources
None.
Disclosures
None.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
| References | ▴Top |
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583-596.
doi pubmed - Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, Sheppard MN, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186-193.
doi pubmed - Fontana M, Pica S, Reant P, Abdel-Gadir A, Treibel TA, Banypersad SM, Maestrini V, et al. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation. 2015;132(16):1570-1579.
doi pubmed - Ruberg FL, Nezafat R. Cardiovascular Magnetic Resonance Visualization of Cardiac Amyloid Infiltration: Challenges and Opportunities. Circulation. 2015;132(16):1525-1527.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.