

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Original Article
Volume 11, Number 6, December 2020, pages 376-385
Sacubitril/Valsartan Inhibits Cardiomyocyte Hypertrophy in Angiotensin II-Induced Hypertensive Mice Independent of a Blood Pressure-Lowering Effect
Kohei Tashiroa, Takashi Kuwanoa, c, Akihito Ideishia, Hidetaka Moritaa, Yoshiaki Idemotoa, Masaki Gotoa, b, Yasunori Suematsua, Shin-ichiro Miuraa, b
aDepartment of Cardiology, Fukuoka University School of Medicine, Fukuoka 814-0180, Japan
bDepartment of Cardiology, Fukuoka University Nishijin Hospital, Fukuoka 814-8522, Japan
cCorresponding Author: Takashi Kuwano, Department of Cardiology, Fukuoka University School of Medicine, 7-45-1 Nanakuma, Jonan-ku, Fukuoka 814-0180, Japan
Manuscript submitted July 18, 2020, accepted August 4, 2020, published online November 2, 2020
Short title: Anti-Hypertrophic Effects of ARNI in Mice
doi: https://doi.org/10.14740/cr1137
| Abstract | ▴Top |
Background: Hypertensive left ventricular hypertrophy is associated with the risk of heart failure, coronary heart disease and cerebrovascular disease. Although sacubitril/valsartan (SAC/VAL), a first-in-class angiotensin receptor neprilysin inhibitor, reduces the risks of death and hospitalization for patients with heart failure, its mechanism of action is not fully understood. We hypothesized that SAC/VAL is superior to other conventional drugs in reducing cardiac hypertrophy.
Methods: Male C57BL/6J mice were implanted with an osmotic pump containing angiotensin II (Ang II). After 7 days of Ang II infusion, mice were also treated with either SAC/VAL, valsartan, enalapril or vehicle alone each day for 2 weeks. Blood pressure measurement was done weekly, and echocardiography was performed before and 3 weeks after infusion of Ang II. Histological analyses were done using extracted heart to investigate cardiac hypertrophy and fibrosis.
Results: Ang II markedly elevated blood pressures in all of the treatment groups, and there were no differences in the degree of blood pressure reduction among the SAC/VAL-, valsartan- and enalapril-treated groups. Echocardiography showed that SAC/VAL significantly suppressed the increase in left ventricular (LV) wall thickness and tended to decrease LV mass. In a histological analysis, SAC/VAL inhibited Ang II-induced cardiomyocyte hypertrophy, and individual cardiomyocytes in the SAC/VAL group were smaller than those in the valsartan and enalapril groups. Although previous studies using animal models of heart failure have indicated that SAC/VAL attenuates cardiac fibrosis, we found no supporting evidence in this setting.
Conclusions: SAC/VAL, valsartan and enalapril all attenuated cardiomyocyte hypertrophy in a mouse model of Ang II-induced cardiac hypertrophy. Of note, SAC/VAL most strongly suppressed hypertrophy in spite of similar blood pressure-lowering effects as valsartan and enalapril. The present study suggests that SAC/VAL may have a beneficial effect on the early stage of hypertensive heart disease.
Keywords: ARNI; Hypertension; Hypertrophy; Myocytes; Sacubitril
| Introduction | ▴Top |
Despite the availability of effective anti-hypertensive drugs, hypertension is still a major risk factor for heart failure, myocardial infarction, stroke and kidney disease [1]. Indeed, more than 40% of patients with hypertension are not controlled even when they receive two or more drugs [2]. Continuous arterial hypertension results in increased cardiac afterload and leads to left ventricular (LV) wall thickening. LV hypertrophy has been associated with increased cardiovascular (CV) risk even though it initially occurs as a compensatory mechanism to minimize wall stress [3]. The renin-angiotensin system (RAS) plays an important role not only in the systemic regulation of arterial pressure and blood volume, but also in regulation of the growth of cardiomyocytes as an autocrine/paracrine factor [4], leading to LV hypertrophy. In fact, chronic infusion of a subpressor dose of angiotensin II (Ang II) into mice induced cardiac hypertrophy and fibrosis [5], and angiotensin-converting enzyme inhibitors (ACEis) and Ang II receptor blockers (ARBs) can reverse LV hypertrophy independent of their effect on blood pressure [6-8]. Natriuretic peptides, including atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP), also counteract RAS activation through their diuretic, vasodilatory and anti-mitogenic properties [9]. Inhibition of neprilysin augments the protective effect against RAS through an increase in natriuretic peptides; thus, simultaneous neprilysin inhibition and angiotensin receptor blockade can theoretically provide beneficial effects against RAS activation. Sacubitril/valsartan (SAC/VAL), a first-in-class angiotensin receptor neprilysin inhibitor (ARNI), has been shown to reduce the risks of death and hospitalization for patients with heart failure [10]. Although patients receiving SAC/VAL had significantly lower blood pressure than those in the ACEi enalapril group in the Prospective Comparison of ARNI with ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) study, it did not affect the differences in event rates. It has also been shown that SAC/VAL leads to sustained reduction in myocardial wall stress and injury in surviving patients with heart failure [11]. These clinical findings suggest that SAC/VAL may reduce LV hypertrophy independent of any lowering of blood pressure. Therefore, we investigated the effect of SAC/VAL on LV remodeling in a mouse model of Ang II-induced cardiac hypertrophy.
| Materials and Methods | ▴Top |
Animal care
Animal experiments were performed according to institutional and governmental guidelines. The protocol was approved by the Animal Care and Use Committee of Fukuoka University. All procedures were in accordance with the Guide for the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources.
Experimental protocol in vivo
Male C57BL/6J mice were purchased from Charles River Laboratories Japan, Inc. (Kanagawa, Japan). Eight-week-old mice were weighed and anesthetized by the inhalation of 1.5% isoflurane. An incision was made in the mid-scapular region, and mini-osmotic pumps (Alzet, CA, USA) were implanted into each animal. The experimental groups received Ang II-loaded pumps that were delivered at a rate of 3.2 mg/kg/day continuously for 3 weeks. Ang II infusion at a rate of 0.5 mg/kg/day is sufficient to increase blood pressure in C57BL6 mice [12], while Ang II is widely used at higher doses (1.4 - 3.2 mg/kg/day) to examine cardiac hypertrophy [13, 14]. Sham-operated animals underwent identical procedures, except that an osmotic pump with saline was implanted. On the seventh day of Ang II infusion, mice were treated with either 60 mg/kg SAC/VAL (salt complex at a 1:1 molar ratio), 30 mg/kg valsartan, or 12mg/kg enalapril dissolved in corn oil, or only corn oil every day for 2 weeks by oral gavage. The dosage of SAC/VAL was determined from previous studies in rodents, all of which used around 60 mg/kg/day of SAC/VAL [15-17]. In addition, it has been reported that drug toxicity was observed at 114 mg/kg/day SAC/VAL [18].
Measurement of blood pressure
Blood pressures were measured by a tail cuff-based MK-2000 (Muromachi Kikai Co., Ltd., Tokyo, Japan) before surgery and before death. Weekly blood pressures were also obtained throughout the experiment to examine the time course of blood pressure elevation in response to Ang II.
Echocardiographic analysis
All mice underwent blinded echocardiography before implantation of the osmotic mini-pump and at the end of the experiment. Echocardiographic analysis was performed using an NEMIO SSA-550A (Toshiba, Tokyo, Japan). Interventricular septum thickness diameter (IVSTd), left ventricular posterior wall thickness diameter (LVPWd), left ventricular internal dimension in diastole (LVDd) and heart rate (HR) were measured in M-mode at the level of the papillary muscle. Echocardiographic LV mass was calculated (mg) as ((left ventricular end-diastolic dimension (LVDEd) + interventricular septal thickness (IVSWTh) + posterior wall thickness (PWTh))3 - LVDd3) × 1.055, where 1.055 (mg/mm3) is the density of the myocardium [19]. LV mass was corrected for body weight and expressed as LV mass index (LVMI).
Histological analysis
At the end of the experiment, all mice were euthanized under anesthesia. The heart tissue was perfused with phosphate-buffered saline (PBS), and then fixed with 4% paraformaldehyde and paraffin embedding. Sliced samples were stained with hematoxylin-eosin and picro-sirius red. For measurement, four random high-power fields from each section were chosen and quantified in a blinded manner using Image J software (National Institutes of Health, MD, USA). The cross-sectional area of individual cardiomyocytes was analyzed quantitatively by morphometry of hematoxylin-eosin stained sections. Four sections for each heart (the mean number of cardiomyocytes was 42 cells per section) were counted. The extent of fibrosis was expressed as the ratio of picro-sirius red stained area to LV wall area.
Quantitative polymerase chain reaction (qPCR) analysis
LV apical regions were frozen in liquid nitrogen and stored at -80 °C. Total RNA was extracted using a RiboPure RNA Purification Kit (Thermo Fisher Scientific Inc., MA, USA). cDNA was produced using a RiverTra Ace qPCR RT Master Mix (TOYOBO Co., Ltd., Osaka, Japan). qPCR was performed with a 7500 Fast Real-Time PCR System (Applied Biosystems, MA, USA) using a THUNDERBIRD SYBR qPCR Mix (TOYOBO, Japan). ANP, BNP and transforming growth factor-β (TGF-β) were investigated. The primers used were forward primer GGGGGTAGGATTGACAGGAT and reverse primer ACACACCACAAGGGCTTAGG for mouse ANP, forward primer TCCTAGCCAGTCTCCAGAGC and reverse primer CCTTGGTCCTTCAAGAGCTG for mouse BNP, and forward primer GCTTCTAGTGCTGACGCCG and reverse primer GACTGGCGAGCCTTAGTTTG for mouse TGF-β.
Statistical analysis
The statistical analysis was performed using SAS software, version 9.4 (SAS Institute, Cary, NC, USA). The values are expressed as the mean and standard deviation. Group differences were analyzed by using the unpaired t-test and the analysis of variance (ANOVA) test. Statistical significance was defined as P value < 0.05.
| Results | ▴Top |
Changes in blood pressure by Ang II infusion
Ang II infusion (3.2 mg/kg/day) gradually increased both systolic and diastolic blood pressures compared with those in the saline-infused sham group, and these increases were significant at 3 weeks (Fig. 1). Mice treated with SAC/VAL (60 mg/kg/day), valsartan (30 mg/kg/day) or enalapril (12 mg/kg/day) had similar levels of blood pressure throughout the experiment. Ang II induced increases in blood pressures in all of the three treatment groups, and the blood pressure values tended to be lower than those in the vehicle-treated control group, although these differences were not significant.
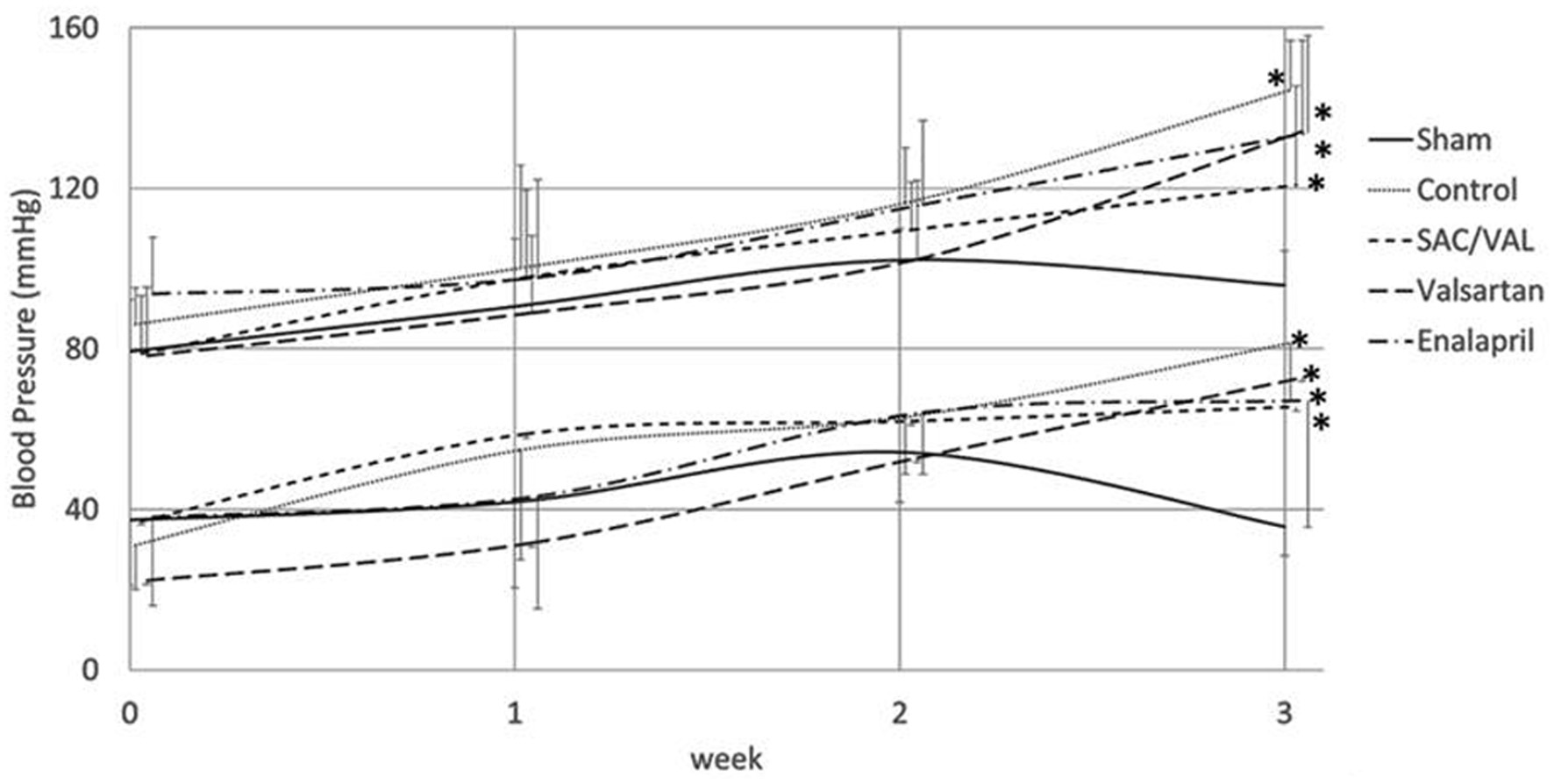 Click for large image | Figure 1. Time course of systolic and diastolic blood pressures. Data are expressed as mean ± SD (n = 6 - 8 each). SD: standard deviation. *P < 0.05 vs. sham-operated group. |
Impact of SAC/VAL treatment on echocardiographic LV hypertrophy in Ang II-induced hypertensive mice
We performed an echocardiographic analysis before Ang II infusion and immediately before we euthanized the animals (Fig. 2). Three weeks of Ang II infusion significantly increased IVSTd and LVPWd, but did not alter LVDd. This suggests that Ang II led to concentric “pathological” hypertrophy [20]. Treatment with SAC/VAL, but not valsartan or enalapril, suppressed the Ang II-induced increase in LV wall thickness. There were also significant differences in IVSTd and LVPWd between the vehicle control group and the SAC/VAL group at the end of the experiment. Ang II infusion increased LV mass, and only treatment with SAC/VAL tended to inhibit this increase (post LVMI; control group 5.6 ± 1.0 mg/g vs. SAC/VAL group 4.7 ± 0.6 mg/g, P = 0.07).
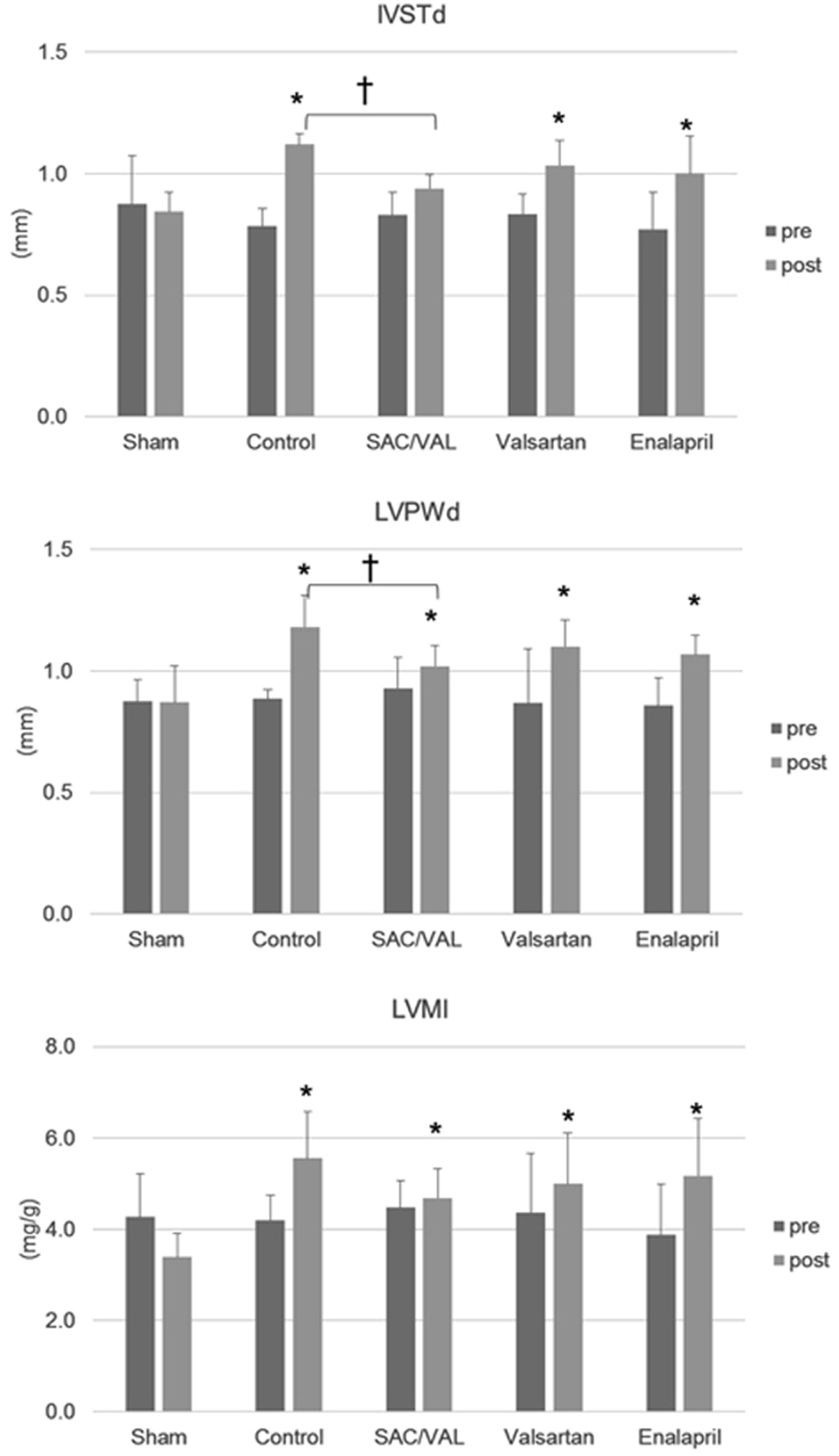 Click for large image | Figure 2. Echocardiographic analysis pre-Ang II infusion and post-3 weeks of Ang II infusion. Data are expressed as mean ± SD (n = 6 - 8 each). IVSTd: interventricular septum thickness diameter; LVPWd: left ventricular posterior wall thickness diameter; LVMI: left ventricular mass index. *P < 0.05 vs. sham-operated group; †P < 0.05. |
Impact of SAC/VAL treatment on cardiomyocyte size and cardiac fibrosis evaluated by a histological analysis
To investigate the effect of SAC/VAL administration on Ang II-induced echocardiographic hypertrophy, we assessed cellular growth, proliferation and fibrosis in the myocardium with hematoxylin-eosin staining and picro-sirius red staining. The mean cross-sectional area of individual cardiomyocytes was enlarged after 3 weeks of Ang II infusion in the vehicle-treated control group (Fig. 3a). The increases in cardiomyocyte size induced by Ang II were significantly attenuated by all three treatments, and the inhibitory effect in the SAC/VAL group was significantly greater than those in the valsartan and enalapril groups (Fig. 3b, the mean area of individual myocytes was 249.5 ± 18.4, 304.8 ± 34.3 and 340.6 ± 41.2 µm2, respectively). In the vast majority of disease processes, cardiac hypertrophy coincides with fibrosis [21]. Therefore, we also examined the extent of cardiac fibrosis by picro-sirius red staining (Fig. 4a). Interstitial and perivascular fibrosis at the mid-transverse section area of the LV wall was modestly increased by Ang II infusion compared to that in sham-treated mice. Remarkably, we found no differences in the extent of fibrosis among the vehicle-control, SAC/VAL, valsartan and enalapril groups (Fig. 4b).
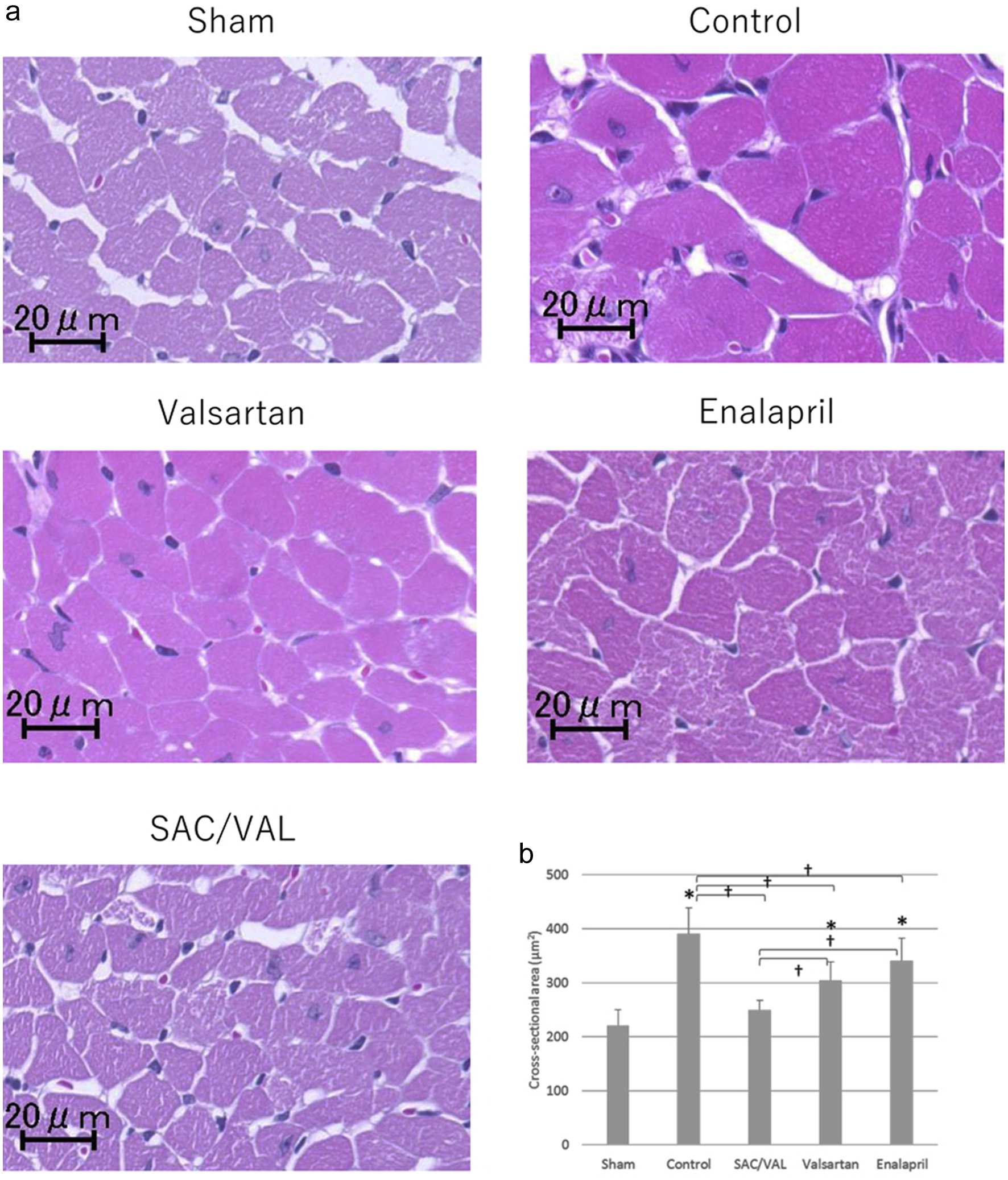 Click for large image | Figure 3. Histological analyses for the cardiomyocytes of left ventricle. (a) Representative photographs of higher magnification views of hematoxylin-eosin-stained heart sections. (b) Quantitative analysis for the cross-sectional area of individual cardiomyocytes. Data are expressed as mean ± SD. SD: standard deviation; SAC/VAL: sacubitril/valsartan-treated group. *P < 0.05 vs. sham-operated group; †P < 0.05 vs. vehicle-treated control group. |
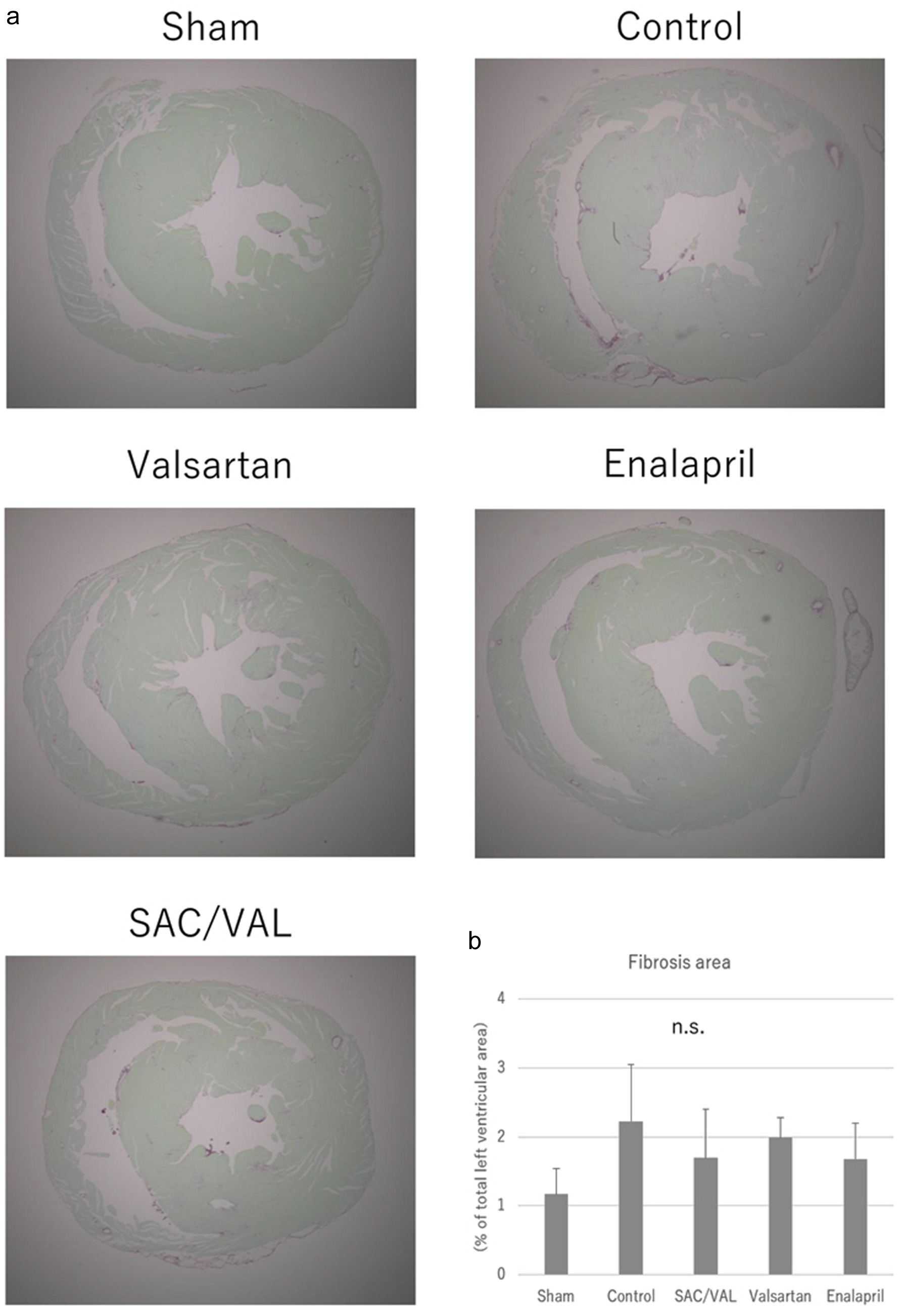 Click for large image | Figure 4. Histological analyses for the cardiac fibrosis. (a) Representative photographs of the picro-sirius red-stained whole heart sections. (b) Quantitative analyses for the left ventricular fibrosis. Data are expressed as mean ± SD. SAC/VAL: sacubitril/valsartan-treated group; SD: standard deviation. |
Effects of SAC/VAL treatment on hypertrophic and profibrotic gene expression induced by Ang II
Transcriptional activation of several genes, including ANP and BNP, is one of the hypertrophic responses to pressure overload [22, 23]. Therefore, we analyzed the mRNA levels of ANP and BNP in the hearts after 3 weeks of Ang II infusion (Fig. 5). Ang II significantly upregulated the expression levels of the ANP gene compared with those in the sham-treated group. The mRNA levels of BNP were also significantly upregulated by Ang II in the vehicle-control, valsartan and enalapril groups, but not in the SAC/VAL group. TGF-β is known as a master regulator of pro-fibrotic signaling, and the mRNA levels of TGF-β were upregulated by Ang II in all of the groups. This is consistent with the finding at the cellular level (Fig. 4) that the expression levels of the pro-fibrotic TGF-β gene were similar among the vehicle-control, SAC/VAL, valsartan and enalapril groups.
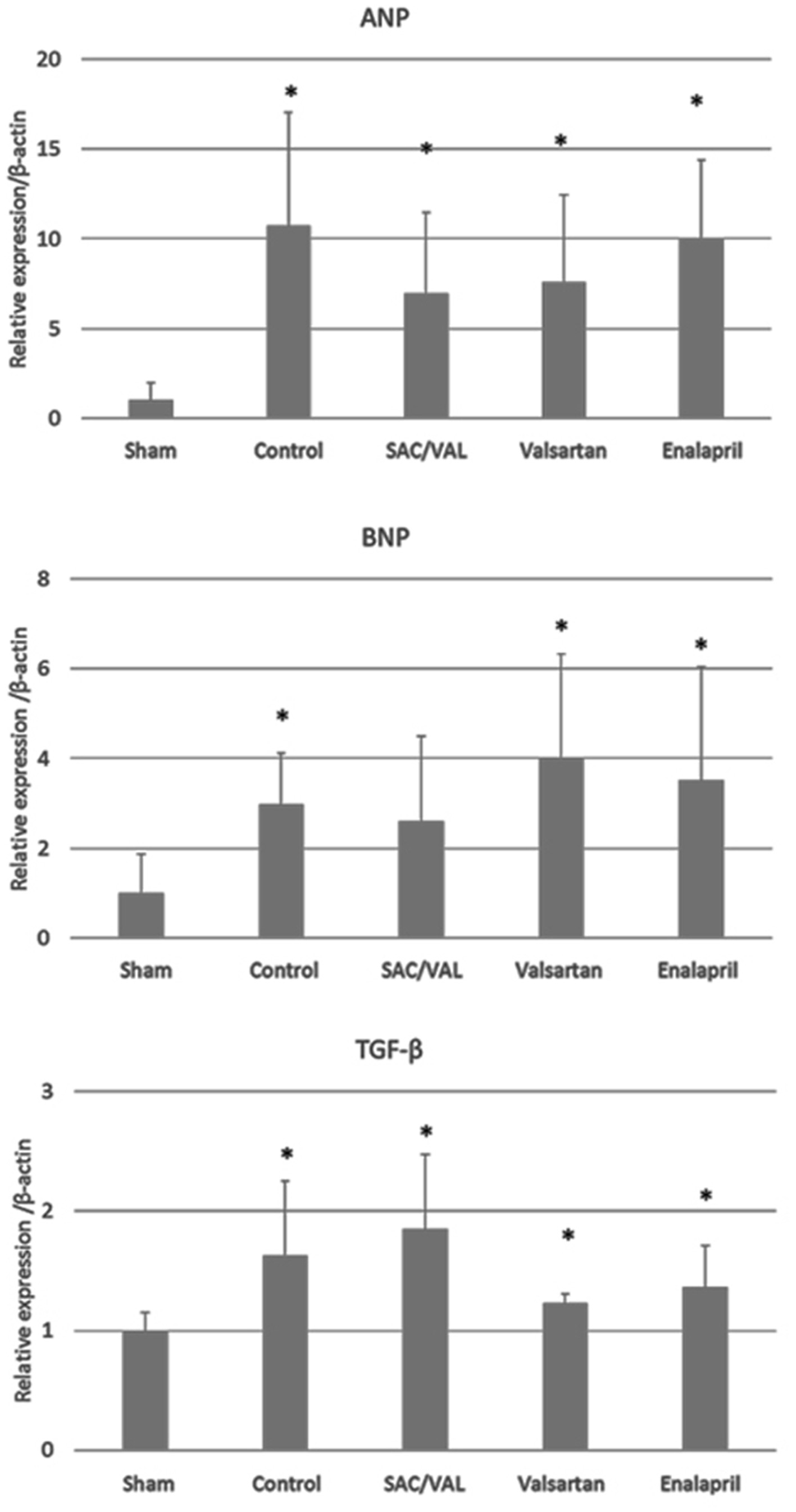 Click for large image | Figure 5. Relative expression of genes of interest by the quantitative polymerase chain reaction. Data are expressed as mean ± SD (n = 6 - 8 each). SD: standard deviation; ANP: atrial natriuretic peptide; BNP: B-type natriuretic peptide; TGF-β: transforming growth factor-β; SAC/VAL: sacubitril/valsartan-treated group. *P < 0.05 vs. sham-operated group. |
| Discussion | ▴Top |
The present study demonstrated that SAC/VAL, a first-in-class ARNI, prevented cardiac hypertrophy in a mouse model of Ang II-induced cardiac hypertrophy. Regardless of similar blood pressure levels, treatment with SAC/VAL significantly inhibited ventricular wall thickening and cardiomyocyte enlargement compared with conventional therapy (ACEi enalapril and ARB valsartan). Furthermore, the anti-hypertrophic effect of SAC/VAL was uncoupled from cardiac fibrosis.
Echocardiographic LV hypertrophy is an independent predictor of CV events, including CV death, coronary events, stroke and heart failure in patients with hypertension [24-26]. Endomyocardial biopsy analysis also showed that cardiomyocyte hypertrophy was an independent predictor of mortality for patients with heart failure, and more than 50% of patients with a cardiomyocyte diameter over 17.7 µm died within 3 years [27]. Meanwhile, meta-analyses reported that LV mass reduction with pharmacological control of blood pressure was associated with a 46% reduction in CV events for hypertension patients [28], and ACEis were the most powerful anti-hypertensive drugs for reducing LV mass, compared to beta blockers, calcium channel blockers and diuretics [29]. In this study, attenuation of cardiac hypertrophy by SAC/VAL was confirmed echocardiographically and histologically, over ACEi or ARB treatment. Therefore, our results support the notion that SAC/VAL has the potential to be the treatment of choice in terms of CV protection for patients with hypertension.
Although the detailed molecular mechanism by which this drug combination confers CV protection is not fully characterized, the present study provides several interesting findings. First, SAC/VAL significantly inhibited LV hypertrophy independent of blood pressure, suggesting a direct rather than indirect effect on cardiac tissue. TGF-β has been proposed to act in an autocrine/paracrine fashion between cardiomyocytes and fibroblasts to stimulate cardiac remodeling [30], and the inhibition of Ang II/TGF-β signaling is a pivotal direct mechanism of the anti-hypertrophic effect of ARBs [31]. In addition, natriuretic peptides also have some direct effects on counteracting RAS signaling [9]. Thus, SAC/VAL may provide an augmented inhibitory effect on ANG II/TGF-β signaling. On the other hand, the concomitant use of ARB with either ACEi or direct renin inhibitor did not further decrease LV hypertrophy, which suggests that simultaneous blocking of RAS signaling for cardiac hypertrophy might reach a plateau [32, 33]. Interestingly, in this study, TGF-β mRNA levels were similar among the three treatment groups, although significant differences were found in cardiac remodeling. This suggests that SAC/VAL might affect LV remodeling not only by blocking Ang II/TGF-β signaling, but also via different pathways. A recent in silico analysis to clarify the details of the mechanism of action of SAC/VAL may support our findings; specifically, sacubitril might attenuate hypertrophy by inhibiting phosphatase and tensin homolog deleted on chromosome ten (PTEN) [34]. In fact, loss of PTEN prevents the development of LV remodeling in response to pressure overload, but not in response to Ang II [35]. Further studies are required to clarify the association between SAC/VAL and PTEN.
Another intriguing finding of this report is that SAC/VAL administration prevented the emergence of hypertrophy, which did not result from the attenuation of cardiac fibrosis. Unlike previous reports in various rodent models of heart failure, which showed that ARNI ameliorates LV remodeling by reducing cardiac fibrosis [15-18], we found significant differences in LV hypertrophy along with a similar degree of cardiac fibrosis among the SAC/VAL, valsartan and enalapril groups. Although 60 mg/kg/day SAC/VAL was not enough to decrease blood pressures in this experiment, the dosing was based on previously published doses of SAC/VAL in rodents. Therefore, this new finding can probably be attributed to the different animal model used. Less interstitial and perivascular cardiac fibrosis (1-2% of the total LV area) was observed in our experiment, perhaps because of the relatively short-term exposure to Ang II. However, validated hypertrophic markers, such as ANP and BNP [23], are significantly upregulated by Ang II infusion, indicating that cardiomyocyte hypertrophy was pathological rather than physiological. Therefore, our results suggest that SAC/VAL may have beneficial effects on the early stage of cardiac remodeling, as well as in patients with chronic stable heart failure with reduced ejection fraction [10]. Meanwhile, the upcoming TRANSITION trial will compare in-hospital initiation of SAC/VAL to post-discharge initiation in patients with acute heart failure occurring as either de novo heart failure or deterioration of chronic heart failure [36], and will hopefully shed some light on the timing for when to initiate SAC/VAL.
In conclusion, SAC/VAL, valsartan and enalapril all attenuated cardiomyocyte hypertrophy induced by Ang II in mice. Of note, SAC/VAL had the strongest inhibitory properties independent of blood pressure-lowering effect and the progression of cardiac fibrosis. To the best of our knowledge, our investigation is the first study to show that SAC/VAL has a beneficial effect on the early stage of hypertensive heart disease.
Acknowledgments
None to declare.
Financial Disclosure
None to declare
Conflict of Interest
None to declare.
Informed Consent
Not applicable.
Author Contributions
KT, TK, and SM conceived and planned the experiments. KT, TK, AI, HM, YI, MG and YS carried out the experiments. KT and TK took the lead in writing the manuscript. SM supervised the project. All authors provided critical feedback and helped shape the research, analysis and manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, Clement DL, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. 2018;39(33):3021-3104.
doi pubmed - Bakris G, Sarafidis P, Agarwal R, Ruilope L. Review of blood pressure control rates and outcomes. J Am Soc Hypertens. 2014;8(2):127-141.
doi pubmed - Muiesan ML, Salvetti M, Monteduro C, Bonzi B, Paini A, Viola S, Poisa P, et al. Left ventricular concentric geometry during treatment adversely affects cardiovascular prognosis in hypertensive patients. Hypertension. 2004;43(4):731-738.
doi pubmed - Lindpaintner K, Ganten D. The cardiac renin-angiotensin system. An appraisal of present experimental and clinical evidence. Circ Res. 1991;68(4):905-921.
doi pubmed - Schultz Jel J, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109(6):787-796.
doi pubmed - Devereux RB, Palmieri V, Sharpe N, De Quattro V, Bella JN, de Simone G, Walker JF, et al. Effects of once-daily angiotensin-converting enzyme inhibition and calcium channel blockade-based antihypertensive treatment regimens on left ventricular hypertrophy and diastolic filling in hypertension: the prospective randomized enalapril study evaluating regression of ventricular enlargement (preserve) trial. Circulation. 2001;104(11):1248-1254.
doi pubmed - Devereux RB, Dahlof B, Gerdts E, Boman K, Nieminen MS, Papademetriou V, Rokkedal J, et al. Regression of hypertensive left ventricular hypertrophy by losartan compared with atenolol: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) trial. Circulation. 2004;110(11):1456-1462.
doi pubmed - Sadoshima J, Izumo S. Molecular characterization of angiotensin II—induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. 1993;73(3):413-423.
doi pubmed - Levin ER, Gardner DG, Samson WK. Natriuretic peptides. N Engl J Med. 1998;339(5):321-328.
doi pubmed - McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371(11):993-1004.
doi pubmed - Packer M, McMurray JJ, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, et al. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation. 2015;131(1):54-61.
doi pubmed - Kawada N, Imai E, Karber A, Welch WJ, Wilcox CS. A mouse model of angiotensin II slow pressor response: role of oxidative stress. J Am Soc Nephrol. 2002;13(12):2860-2868.
doi pubmed - Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, et al. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004;10(12):1384-1389.
doi pubmed - Wang HD, Xu S, Johns DG, Du Y, Quinn MT, Cayatte AJ, Cohen RA. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ Res. 2001;88(9):947-953.
doi pubmed - von Lueder TG, Wang BH, Kompa AR, Huang L, Webb R, Jordaan P, Atar D, et al. Angiotensin receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail. 2015;8(1):71-78.
doi pubmed - Suematsu Y, Miura S, Goto M, Matsuo Y, Arimura T, Kuwano T, Imaizumi S, et al. LCZ696, an angiotensin receptor-neprilysin inhibitor, improves cardiac function with the attenuation of fibrosis in heart failure with reduced ejection fraction in streptozotocin-induced diabetic mice. Eur J Heart Fail. 2016;18(4):386-393.
doi pubmed - Kusaka H, Sueta D, Koibuchi N, Hasegawa Y, Nakagawa T, Lin B, Ogawa H, et al. LCZ696, angiotensin II receptor-neprilysin inhibitor, ameliorates high-salt-induced hypertension and cardiovascular injury more than valsartan alone. Am J Hypertens. 2015;28(12):1409-1417.
doi pubmed - Burke RM, Lighthouse JK, Mickelsen DM, Small EM. Sacubitril/Valsartan decreases cardiac fibrosis in left ventricle pressure overload by restoring PKG signaling in cardiac fibroblasts. Circ Heart Fail. 2019;12(4):e005565.
doi - Gardin JM, Siri FM, Kitsis RN, Edwards JG, Leinwand LA. Echocardiographic assessment of left ventricular mass and systolic function in mice. Circ Res. 1995;76(5):907-914.
doi pubmed - Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123(3):327-334.
doi pubmed - Ho CY, Lopez B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, et al. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010;363(6):552-563.
doi pubmed - Komuro I, Yazaki Y. Control of cardiac gene expression by mechanical stress. Annu Rev Physiol. 1993;55:55-75.
doi pubmed - Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14(1):38-48.
doi pubmed - Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322(22):1561-1566.
doi pubmed - Bluemke DA, Kronmal RA, Lima JA, Liu K, Olson J, Burke GL, Folsom AR. The relationship of left ventricular mass and geometry to incident cardiovascular events: the MESA (Multi-Ethnic Study of Atherosclerosis) study. J Am Coll Cardiol. 2008;52(25):2148-2155.
doi pubmed - Gardin JM, McClelland R, Kitzman D, Lima JA, Bommer W, Klopfenstein HS, Wong ND, et al. M-mode echocardiographic predictors of six- to seven-year incidence of coronary heart disease, stroke, congestive heart failure, and mortality in an elderly cohort (the Cardiovascular Health Study). Am J Cardiol. 2001;87(9):1051-1057.
doi - Vigliano CA, Cabeza Meckert PM, Diez M, Favaloro LE, Cortes C, Fazzi L, Favaloro RR, et al. Cardiomyocyte hypertrophy, oncosis, and autophagic vacuolization predict mortality in idiopathic dilated cardiomyopathy with advanced heart failure. J Am Coll Cardiol. 2011;57(14):1523-1531.
doi pubmed - Pierdomenico SD, Cuccurullo F. Risk reduction after regression of echocardiographic left ventricular hypertrophy in hypertension: a meta-analysis. Am J Hypertens. 2010;23(8):876-881.
doi pubmed - Dahlof B, Pennert K, Hansson L. Reversal of left ventricular hypertrophy in hypertensive patients. A metaanalysis of 109 treatment studies. Am J Hypertens. 1992;5(2):95-110.
doi pubmed - Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106(11):1675-1680.
doi pubmed - Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J Clin Invest. 2010;120(10):3520-3529.
doi pubmed - Makani H, Bangalore S, Desouza KA, Shah A, Messerli FH. Efficacy and safety of dual blockade of the renin-angiotensin system: meta-analysis of randomised trials. BMJ. 2013;346:f360.
doi pubmed - Solomon SD, Appelbaum E, Manning WJ, Verma A, Berglund T, Lukashevich V, Cherif Papst C, et al. Effect of the direct Renin inhibitor aliskiren, the Angiotensin receptor blocker losartan, or both on left ventricular mass in patients with hypertension and left ventricular hypertrophy. Circulation. 2009;119(4):530-537.
doi pubmed - Iborra-Egea O, Galvez-Monton C, Roura S, Perea-Gil I, Prat-Vidal C, Soler-Botija C, Bayes-Genis A. Mechanisms of action of sacubitril/valsartan on cardiac remodeling: a systems biology approach. NPJ Syst Biol Appl. 2017;3:12.
doi pubmed - Oudit GY, Kassiri Z, Zhou J, Liu QC, Liu PP, Backx PH, Dawood F, et al. Loss of PTEN attenuates the development of pathological hypertrophy and heart failure in response to biomechanical stress. Cardiovasc Res. 2008;78(3):505-514.
doi pubmed - Pascual-Figal D, Wachter R, Senni M, Belohlavek J, Noe A, Carr D, Butylin D. Rationale and design of TRANSITION: a randomized trial of pre-discharge vs. post-discharge initiation of sacubitril/valsartan. ESC Heart Fail. 2018;5(2):327-336.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.