

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Original Article
Volume 12, Number 1, February 2021, pages 16-24
Some Aspects of Role of Nitric Oxide in the Mechanisms of Hypertension (Experimental Study)
Katevan Kakabadzea, Irakli Megreladzeb, Nino Khvichiac, Nodar Mitagvariad, Nina Kipiania, Megi Dumbadzee, Tamar Sanikidzea, d, f
aDepartment of Physics, Biophysics, Biomechanics and Informational Technologies, Tbilisi State Medical University, Tbilisi, Georgia
bDepartment of Cardiology, Amtel Hospital, Tbilisi, Georgia
cDepartment of Pathology, Faculty of Medicine, I. Javakhishvili Tbilisi State University, Tbilisi, Georgia
dI. Beritashvili Center of Experimental Biomedicine, Tbilisi, Georgia
eEuropean University, Tbilisi, Georgia
fCorresponding Author: Tamar Sanikidze, Department of Physics, Biophysics, Biomechanics and Informational Technologies, Tbilisi State Medical University, 33 Vaja Pshavela Av., Tbilisi 380060, Georgia
Manuscript submitted September 22, 2020, accepted November 18, 2020, published online December 11, 2020
Short title: Role of NO in the Mechanisms of Hypertension
doi: https://doi.org/10.14740/cr1172
| Abstract | ▴Top |
Background: Modulation of endothelial function is a therapeutic option to reduce some of the significant complications of hypertension. However, the relationship between endothelial dysfunction reduced nitric oxide (NO) production, and the development of hypertension is not fully understood. To establish a potential pathogenetic link between impaired NO synthesis and hypertension, we investigated the results of competitive interaction of the substrate of NO synthase, L-arginine, and its analog, an non-selective inhibitor of NO synthase, N-nitro-methyl ether-L-arginine (L-NAME), in experimental rats.
Methods: Arterial hypertension was induced in male Wistar rats by intraperitoneal administration of L-NAME (Sigma-Aldrich) for 4 - 7 weeks. During the last 3 weeks, to a separate group of animals simultaneously with L-NAME, L-arginine (Sigma-Aldrich) was administered. In animals monitored for systolic and diastolic pressure, the level of NO in blood samples was determined spectrophotometrically using a Griess reagent.
Results: Administration of L-NAME for 4 - 7 weeks induced an irreversible decrease of NO content in blood, a reversible increase of systolic pressure (SP) and diastolic pressure (DP), and an irreversible increase in pulse pressure (PP). In rats against the background of 7 weeks of intraperitoneal administration of L-NAME, during the last 3 weeks, they were injected with L-arginine, the SP and DP indices returned to their initial values, PP decreased and the NO content in arterial blood increased.
Conclusions: The results of the study indicate the presence of residual endothelial dysfunction (characterized by insufficient NO) after the correction of hypertension. Therefore, in developing the new therapeutic approaches for the treatment of hypertension, it is necessary to include drugs that, in addition to correcting blood pressure, will support normalization, and potentiation of endothelial function and endogenous NO synthesis.
Keywords: Nitric oxide; Hypertension; Endothelial dysfunction; Systolic pressure; Diastolic pressure; Pulse pressure
| Introduction | ▴Top |
The etiology of hypertension and its relationship with disorders of nitric oxide (NO) production, the question of whether dysfunction of the endothelium and a decrease in NO production occurs before or after the development of hypertension, remains unclear. Since endothelial dysfunction is usually observed in various types of hypertension and can be restored by correcting blood pressure, it is believed that the extrusion of endothelial function is not a primary cause, but a secondary result of hypertension. However, impaired NO-dependent vasodilation has been shown to precede hypertension in black patients with normotension and in normotensive offsprings of hypertensive parents [1]. Although, due to the modest percentage of the heritability of endothelial dysfunction, measured as flow-mediated dilation of the brachial artery, generally, it is difficult to assert that endothelial dysfunction is a cause of hypertension [2, 3].
The theory of “endothelial dysfunction”, and consequential insufficient NO production in human with essential hypertension led to the creation of an animal model of human hypertension where NO deficiency, achieved by both acute and chronic inhibition of NO synthase (NOS) with arginine derivatives, induced a significant rise in blood pressure (so-called “NO-deficient hypertension” in normotensive rats) [4], and this model became widely used for investigation of the NO participation in cardiovascular disorders [5].
The mechanism of NOS inhibition by substrate analogs consists of competitive binding to the enzyme. Reversible inhibitor and one of the most frequently used L-arginine substituents is N-nitro-methyl ether-L-arginine (L-NAME), which is considered to be a non-selective inhibitor of NOS [6-9] (a competitive inhibitor of all NOSs having higher selectivity to endothelial NOS (eNOS) and neuronal NOS (nNOS) over inducible NOS (iNOS) [10]). Administration of L-NAME is also associated with increased production of reactive oxygen species (ROS) accompanied by depletion of endogenous antioxidants [11, 12]. Under conditions of intensified intracellular ROS production, oxidation of the tetrahydrobiopterin and other cofactors needed for NO synthesis leads to the uncoupling of the NOS dimer which results in decreased NOS activity. Additionally, in the condition of oxidative stress, NOS produces rather superoxide radical than NO, and oxidative degradation of NO to peroxynitrite is also possible. Therefore, chronic administration of L-NAME in experimental rats is accompanied by a significant decrease in the synthesis of NO.
Modulation of endothelial function is a therapeutic option to reduce some of the significant complications of hypertension. However, the relationship between endothelial dysfunction reduced NO production, and the development of hypertension is not fully understood.
To establish physiological and pathophysiological aspects of NO role in the mechanisms of hypertension, a potential pathogenetic link between impaired NO synthesis and endothelial dysfunction, we investigated the competitive interaction of the substrate of NOS, L-arginine and its analog L-NAME in experimental rats.
| Materials and Methods | ▴Top |
Animal models of hypertension
In the study, 13- to 14-month-old (body weight 200 - 220 g) white Wistar male rats (35 animals) were used. All animals were kept in acrylic cages with wood shavings in an acclimatized room (12/12 h light/dark cycle; 22 ± 3 °C) with free access to food and water. All animal procedures were approved by the Animal Care and Use Committee of the Tbilisi State Medical University and were conducted in accordance with the “Guide for the Care and Use of Laboratory Animals” (NIH Publication No. 85-23, revised 1996). All efforts were made to minimize the number of animals and their suffering throughout the experiment.
Hypertension was induced with intraperitoneal administration of L-NAME (Sigma-Aldrich) (40 mg/kg).
The animals were divided into three groups. 1) Group I, control group (seven intact rats). 2) Group II, experimental group (an experimental model of hypertension (21 rats) contained three (IIa, IIb and IIc) subgroups (seven rats in each subgroup)): IIa subgroup - intraperitoneal administration of L-NAME (40 mg/kg) during 4 weeks; IIb subgroup - intraperitoneal administration of L-NAME (40 mg/kg) during 7 weeks; IIc subgroup - intraperitoneal administration of L-NAME (40 mg/kg) during 4 weeks followed by 3 weeks of spontaneous recovery. 3) Group III, experimental group (seven rats) (intraperitoneal administration of L-NAME (40 mg/kg) for 7 weeks, and from the beginning of the fifth week (weeks 5-7), intraperitoneal administration of L-arginine (Sigma-Aldrich) (300 mg/kg) was added).
In control (group I) and experimental (group II (subgroups IIa, IIb and IIc) and group III) rats, parallel blood samples were drawn from the radial artery and brachial vein under anesthesia with 2% ether. At the end of the acute experiment, the chest was opened, and the middle part of the carotid artery was excised for morphological studies.
Blood pressure measurement
The blood pressure (systolic pressure (SP) and diastolic pressure (DP)) of the rats was measured every second day by the tail-cuff method by equipment “Systola” and oscillograph (“Neurobotics” LLC, Russia) according to the manufacturer’s protocol.
Measurement of total NO level in blood
The level of NOx in blood samples was determined by a modified method of Miranda et al [13]. As the first step, blood serum sample deproteinization was achieved by adding equal volumes of 0.3 M NaOH to 100 µL of blood serum. It was mixed well and incubated for 5 min at room temperature. Then 100 µL of 5% ZnSO4 was added, mixed well and incubated for additional 5 min at room temperature. After the incubation, the mixture was centrifuged at 3,000 rpm at 4 °C for 15 min. An aliquot of 100 µL of the clear supernatant was then mixed with 200 µL of Griess reagent.
Griess reagent was prepared just prior to the assay and contained 0.25% VCl3, 0.1% sulfanilamide and 0.05% N-(1-naphthyl)-ethylenediamine (NED) (Sigma-Aldrich) in 0.5 M HCl. Reagent blank was the same but contained 100 µL of distilled water instead of the blood serum sample. The mixture was incubated for 30 min at 37 °C and absorbance was measured at 540 nm with a microplate reader (Multiclan GO, Thermo Fisher Scientific, Finland). The standard curve for NaNO2 was used to calculate total NOx concentration in the samples [13].
Morphological study
The carotid artery was cleaned and divided into proximal 1 mm long segments and immersed into the fixative (glutaraldehyde 3% in 0.1 M phosphate buffer) for a further 3 h; after washing in a phosphate buffer, the segments were fixed with 2% OsO4 in 0.1 M phosphate buffer. Then the specimens were stained with 2% uranyl acetate, dehydrated through ascending concentrations of alcohol and embedded in Durcupan ACM. Three randomly selected segments of the coronary artery were cut perpendicularly to the long axis. Both the inner circumference and arterial wall thickness (WT) were measured under light microscopy. The inner diameter (ID) and the WT (tunica intima and tunica media) of the carotid artery were calculated.
Statistical analysis
Statistical analysis of obtained results was performed by the use of the SPSS statistical analysis program package (version 10.0). The average parameters and their statistical derivations were analyzed. The difference between groups was evaluated by Student’s t-test. In all cases, statistical significance was obtained at P < 0.05. To establish the relationship between changes in blood NOx and pulse pressure (PP), Pearson’s correlation coefficients were calculated.
| Results | ▴Top |
L-NAME administration results
After 4 - 7 weeks of intraperitoneal administration of L-NAME (40 mg/kg) to rats (subgroups IIa and IIb), SP and DP increased by 81% and by 76%. Three weeks after the discontinuation of the 4-week intraperitoneal administration of L-NAME (spontaneous recovery), the indicators of SP and DP returned to the initial level (subgroup IIc) (Fig. 1). This means that L-NAME-induced experimental hypertension is reversible.
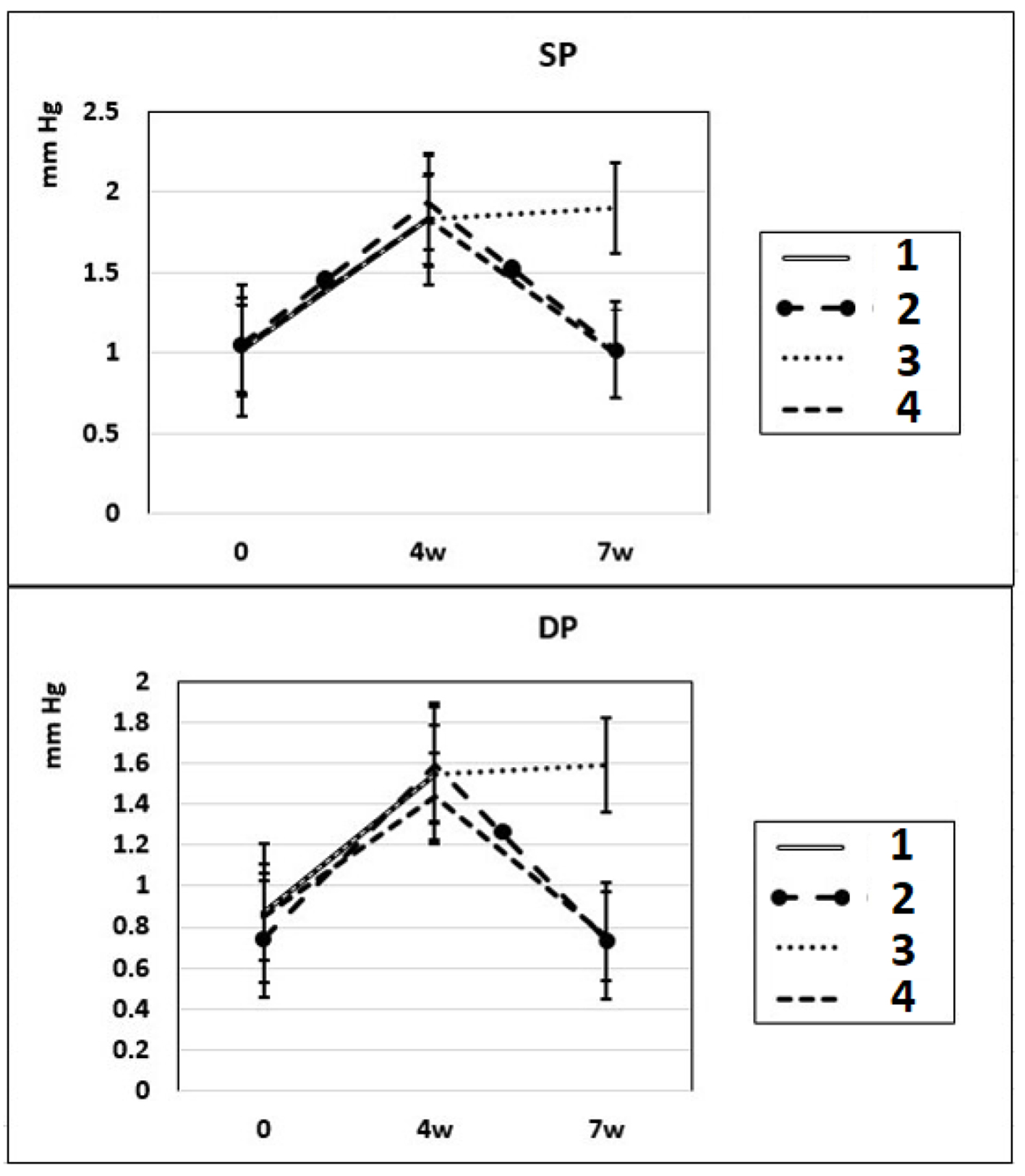 Click for large image | Figure 1. Dynamics of alterations in SP and DP in experimental rats. 1: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 4 weeks; 2: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 4 weeks followed by 3 weeks of spontaneous recovery; 3: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 7 weeks (4 + 3 weeks); 4: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 7 weeks, from the beginning of the fifth week (weeks 5 - 7), intraperitoneal administration of L-arginine (300 mg/kg) was added. SP: systolic pressure; DP: diastolic pressure; L-NAME: N-nitro-methyl ether-L-arginine. |
In these experimental groups, the pulse rate had a tendency to increase (statistically insignificant) during 4 weeks of L-NAME administration (subgroup IIa) and stay at this level for the next 3 weeks of spontaneous recovery (subgroup IIc); after 7 weeks, pulse rate statistically significantly increased by 3% (P < 0.005) compared to the initial level (subgroup IIb). PP (the difference between SP and DP (SP - DP)) increased by 72% and 93% in subgroups IIa and IIb, respectively, and remained at this level after 3 weeks of discontinuation of L-NAME administration in rats (subgroup IIc) (Fig. 2). These data indicate that after 4 weeks of exposure to L-NAME, rats develop irreversible changes in vascular elasticity (stiffening).
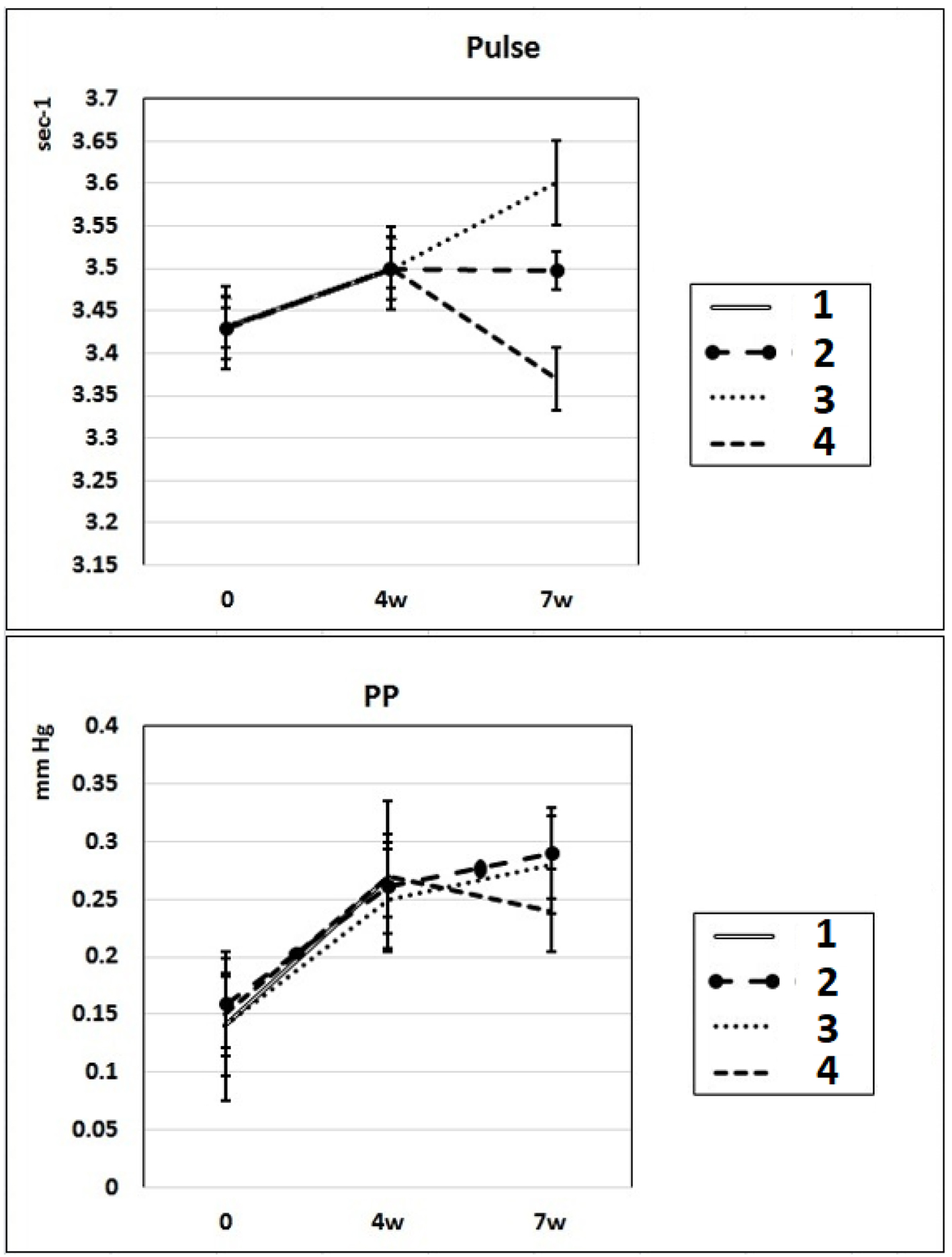 Click for large image | Figure 2. Dynamics of alterations of indicators of pulse rate and PP in experimental rats. 1: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 4 weeks; 2: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 4 weeks followed by 3 weeks of spontaneous recovery; 3: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 7 weeks (4 + 3 weeks); 4: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 7 weeks, from the beginning of the fifth week (weeks 5 - 7), intraperitoneal administration of L-arginine (300 mg/kg) was added. PP: pulse pressure; L-NAME: N-nitro-methyl ether-L-arginine. |
In subgroup IIa (L-NAME administered for 4 weeks), the NO content decreased by 30% in the arterial blood and by 23% in the venous blood of rats compared to baseline. After 7 weeks of L-NAME treatment (subgroup IIb), the NO content in the arterial blood continued to decrease (by 18%) (Fig. 3). In subgroup IIc (3 weeks after discontinuation of 4 weeks of L-NAME administration), the NO content did not differ from the control level in either arterial or venous blood (Fig. 3).
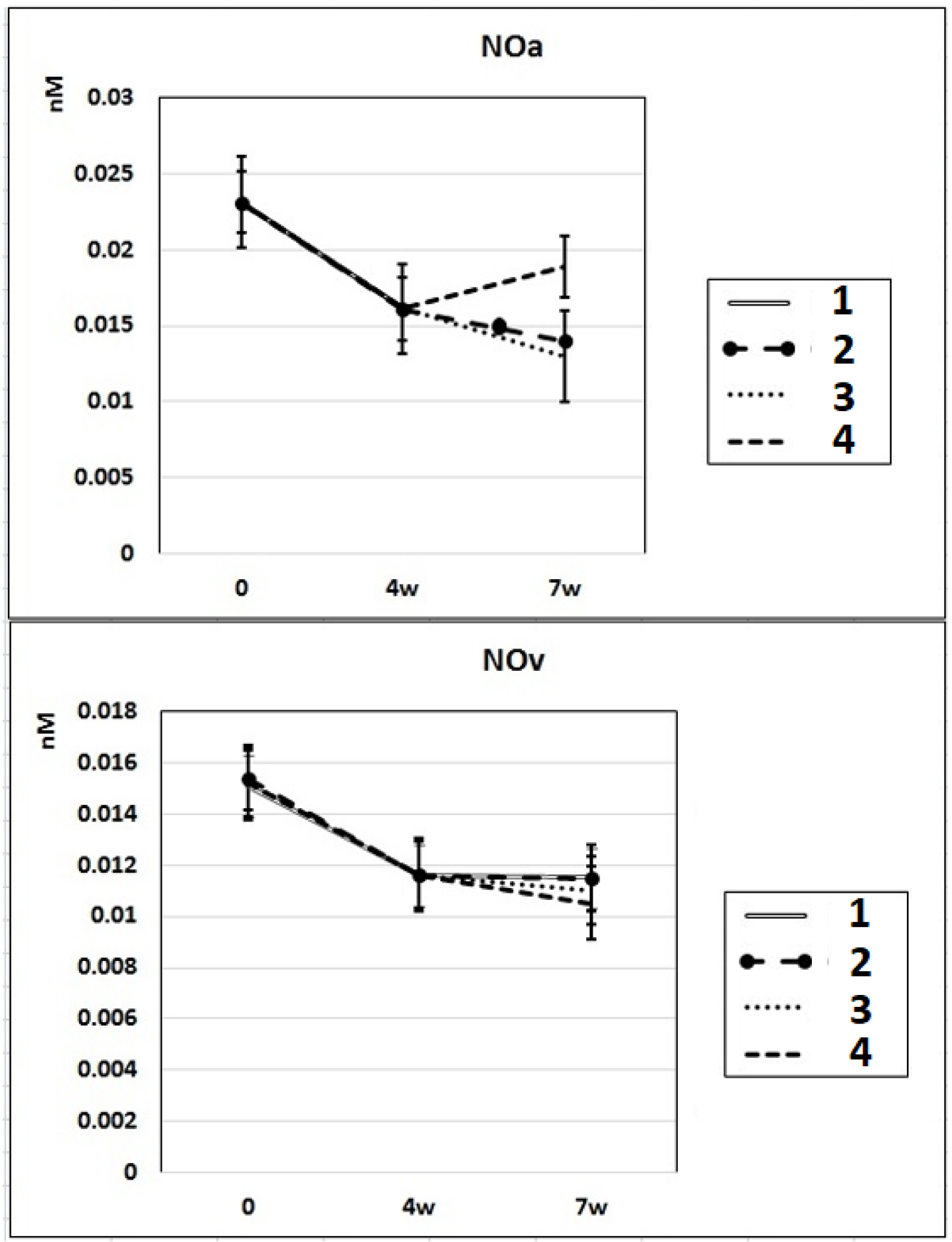 Click for large image | Figure 3. Dynamics of alterations of NO content in the rats’ arterial and venous blood. 1: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 4 weeks; 2: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 4 weeks followed by 3 weeks of spontaneous recovery; 3: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 7 weeks (4 + 3 weeks); 4: rats with intraperitoneal administration of L-NAME (40 mg/kg) for 7 weeks, from the beginning of the fifth week (weeks 5 - 7), intraperitoneal administration of L-arginine (300 mg/kg) was added. NO: nitric oxide; L-NAME: N-nitro-methyl ether-L-arginine. |
L-arginine treatment results of L-NAME-induced hypertension
In rats of group III, where, against the background of 7 weeks of intraperitoneal administration of L-NAME (40 mg/kg) starting from the fifth week (weeks 5 - 7) for 3 weeks, they were treated with L-arginine (300 mg/kg), at the end of the experiment, the SP and DP indices returned to the initial level (Fig. 1), and the NO content in arterial blood increased by 29% compared with the value in animals untreated with L-arginine (Fig. 3). These data indicate that the combined administration of exogenous L-arginine with L-NAME provides restoration of NOS activity, which in turn contributes to an increase in the level of NO in arterial blood. In this experimental group (group III), the PP decreased by 18% compared with the values characteristic for subgroup IIb, which indicates an increase in the elasticity of the arterial walls (Fig. 2).
Morphological study results
The results of the morphometric evaluation of the geometry of the carotid artery showed that in control animals, the ID of the carotid artery was 825 ± 25.5 µm, and no significant alterations were found in the ID after treatment with L-NAME (subgroups IIa, IIb and IIc), or L-NAME plus L-arginine (group III) (Table 1). The WT (tunica intima + tunica media) of the carotid artery in control (group I) was 20.09 ± 0.85 µm; in animals treated with L-NAME (subgroups IIa, IIb and IIc), it was increased by 41-57%; after the additional treatment with L-arginine (group III), the WT of the carotid artery decreased by 27%.
 Click to view | Table 1. Morphometric Parameters of Carotid Artery |
| Discussion | ▴Top |
NO regulates cardiac function through both vascular-dependent (regulation of coronary vessel tone, thrombogenicity, and proliferative and inflammatory properties as well as cellular interactions supporting angiogenesis) and independent effects (the direct effects of NO on cardiomyocyte contractility (fine regulation of excitation-contraction coupling to modulation of autonomic signaling (presynaptic and postsynaptic) and mitochondrial respiration). This multifaceted involvement of NO is carried out by complex molecular mechanisms of regulation of NO synthesis. Disruption of these specific mechanisms or suppression of NOS activity can lead to profound cellular disorders leading to heart failure [14]. Chronic treatment with L-NAME commonly is used for the induction of NO-deficient hypertension. In our experiments, chronic administration of L-NAME for 4 - 7 weeks induced a decrease of NO content in rats’ arterial and venous blood compared to baseline (Fig. 3), which resulted in an increase of SP, DP and PP compared to control.
In some experiments, after prolonged administration of a low dose of L-NAME, paradoxically increased NOS activity in peripheral tissues, raised endothelium-dependent vasorelaxation and decreased arterial vasoconstriction were detected, which indicates that chronic treatment with low doses of L-NAME may cause animals adaptation and reduce the likelihood or prevent the development of hypertension at all [5, 15]. The mechanism of L-NAME-induced adaptation involves activation of the transcriptional regulatory protein, nuclear factor-kB (NF-kB), caused by both a decrease in NO level and an increase in ROS generation [16], which is usually associated with an increase in eNOS and iNOS expression [17-20]. In our experiments, we used a relatively high dose of L-NAME (40 mg/kg) which ensured the blockage of NOS activity even in case of its possible compensatory excessive expression. According results of our study under 4 - 7 week administration of L-NAME, NOx (nitrite + nitrate) content decreased by 31-40% in arterial blood (0.023 ± 0.003 nM (control), 0.016 ± 0.003 nM (after 4 weeks) and 0.014 ± 0.002 nM (after 7 week)) and by 20-27% in vein blood (0.015 ± 0.0015 nM (control), 0.012 ± 0.0015 nM (after 4 weeks) and 0.011 ± 0.002 nM (after 7 week)) (Fig. 3). Our indicators of NOx content in arterial blood (NOxa) coincide with literature data of the average level of NOx in peripheral blood [21]. Literature data strongly suggest that in vivo the endothelial release of NO in arteries and veins is different [22-24]. NO production is thought to occur mainly in the arterial side of the circulation. The continuous (basal) as well as induced NO release from the arterial endothelium has been clearly demonstrated both in preparations of isolated arteries and in perfused arterial vascular beds [22, 25]. Isolated preparations of veins showed much less endothelium-dependent relaxation evidence compared to that in arteries [25]. In addition, infusion of the potent inhibitor of NO formation, L-NAME, into the brachial artery induces direct vasoconstriction, but it has no such direct effect on the veins of hand [22, 24]. The venous plasma levels of nitrite are known significantly lower than in arterial plasma (which is consistent with the results of our experiments - NO content in arterial blood was 50% higher than in venous blood) due to the high lability of NO (half-life of a few seconds), which degrades to stable metabolites, nitrite and nitrate [23, 24]. The arterial endothelium plays a critical role in controlling the vascular tone.
The results of our experiments show that 3 weeks after 4 weeks-lasted L-NAME administration (subgroup IIIc), L-NAME-induced increase of SP and DP was reversible and partially normalized; however, the level of NO in the blood remained low, and PP - an indicator of large artery stiffness [26], was high. In these experimental groups, the pulse rate during 4 weeks of L-NAME administration (subgroup IIa) had a tendency to increase (statistically insignificant) and stay at this level for the next 3 weeks of spontaneous recovery (subgroup IIc); after 7 weeks of L-NAME administration (subgroup IIb), the pulse rate statistically significantly increased by 3% (P < 0.005) compared to the initial level.
Freshly dissolved L-NAME contained 2% of L-NOARG and was hydrolyzed with a half-life of 365 ± 11.2 min in buffer (pH 7.4), 207 ± 1.7 min in human plasma and 29 ± 2.2 min in whole blood [27]. In our experiments, L-NAME lasted for a long time (4 - 7 weeks) that ensured permanent blocking of NOS activity.
A large number of studies indicate the negative effects of long-term treatment with L-NAME on cardiac function included myocardial hypertrophy and fibrosis [28, 29], decreased cardiac output, lowered regional blood flow, suppressed angiogenesis by growth factors stimulation [30, 31], reduced vasorelaxation as well as vascular wall thickening [32, 33] and finally, an increase in mortality in several cases. Disorders in the heart can naturally increase the pulse rate, to compensate for any loss of contractility or ejection fraction, which was manifested in our experiments.
An irreversible increase in PP after 4 - 7 week of L-NAME treatment indicates the irreversible character of the alterations of vascular elasticity (stiffening) [34], a decrease in their compliance due to the structural molecular changes in the arterial wall, including decreased elastin content, increased collagen I deposition and calcification. Literature data show that 3 - 4 weeks following L-NAME treatment contribute vascular lesions, such as perivascular fibrosis, medial thickening and increase in intima/media or wall/lumen ratios in the mesenteric microvascular beds, arterioles, coronary arteries and aorta [35]. Elevated stiffness of large arteries along with the increase of carotid-intima media thickness, and the impaired flow-mediated dilatation, are considered markers of endothelial dysfunction [36]. After removal of vascular endothelium in animals, the stiffness of large arteries alters by a factor of 5.6, suggesting that substances derived from endothelium regulate arterial stiffness in vivo. The endothelium releases a number of mediators including NO, which can alter smooth muscle tone, smooth muscle proliferation, participates in the regulation of the compliance of arteries walls through modification of the tropoelastin gene expression [37], inhibits mineralization and differentiation of the vascular smooth muscle cells (VSMCs) into osteoblastic cells responsible for the vascular calcification [38]. Endothelial dysfunction, characterized by decreased bioavailability of NO, is a predictor of cardiovascular risk and outcome, and therapeutic interventions that improve endothelial function also reduce arterial stiffness, which suggests that NO may itself regulate large arterial stiffness [26].
In the case of the simultaneous action of an inhibitor (L-NAME) and a substrate (L-arginine) of NOS, the significant increase of the NO content in arterial blood (but not in venous) was found (Fig. 3). SP, DP and PP indicators also returned to the initial level. These data indicate that the competitive substitution of L-NAME with L-arginine contributes to the restoration of NOS activity, increasing the level of NO in arterial blood, normalization of arterial pressure, restoration of endothelial function and blood vessel elasticity. Since in the case of a 3-week respite after 4-week administration of L-NAME against the background of partial normalization of SP and DP, the level of NO in arterial blood remained low, and PP remained high; it seems that endogenous reserves of arginine are not sufficient to complete restoration of blood vessels elasticity and vasodilation. Additional administration of exogenous L-arginine during the last 3 weeks of L-NAME administration (we administered a dose of L-arginine that is almost two times higher than normal cellular and plasma levels of L-arginine (100 - 200 µM)) not only annihilated the inhibitory effect of L-NAME, but also provided normalization of NO synthesis and restoration of vascular elasticity (decrease in PP), which means normalization of NO-dependent endothelial function. These data are convincing evidence in favor of the important role of NO in the normalization of the studied parameters. Endothelial dysfunction, characterized by decreased bioavailability of NO, is a predictor of cardiovascular risk and outcome and several therapeutic interventions that improve endothelial function also reduce arterial stiffness, suggesting that NO may itself regulate large arterial stiffness [26].
Correlation analysis results show that the overall correlation and its statistical significance between NO and PP levels (the combined group including parameters of subgroups I, IIa and IIb) are quite high (Fig. 4), while the correlation and its statistical significance between NO and PP within groups (I, IIa, IIb, IIc and III) are quite low (Table 2). This suggests that the overall correlation is completely due to between-groups (I, IIa, IIb, IIc and III) correlations, and indicates an indirect causal relationship between NO and PP levels.
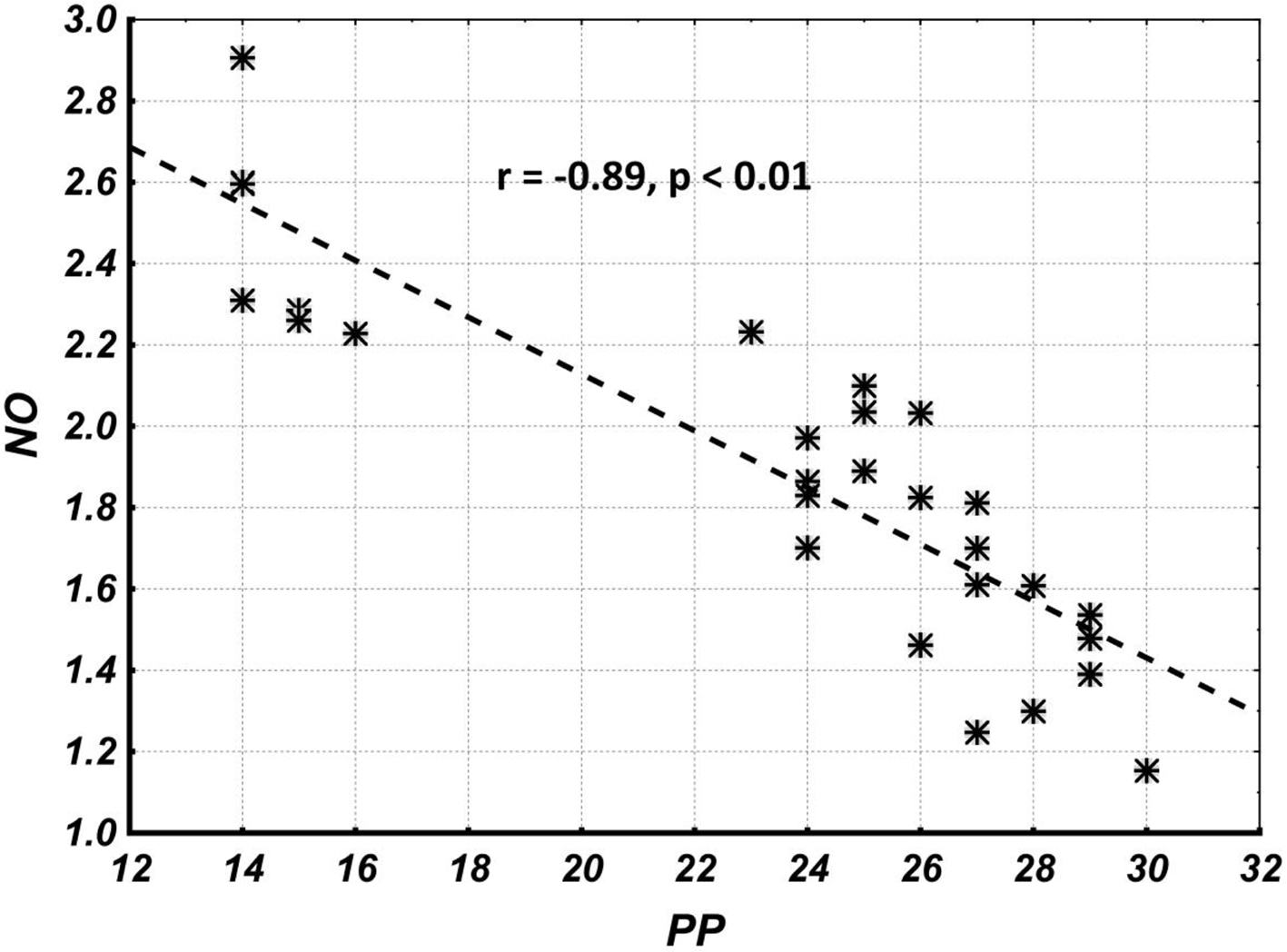 Click for large image | Figure 4. The overall correlation and its statistical significance between NO and PP levels (combined group including parameters of subgroups I, IIa, IIb and IIc). NO: nitric oxide; PP: pulse pressure. |
Click to view | Table 2. Correlation and Its Statistical Significance Between NO and PP Levels Within Groups |
In addition, results of the morphometric assessment of the carotid artery in animals treated with L-NAME for 4 - 7 weeks show an increase in its WT (tunica intima + tunica media) by 41-57% (without changing inner diameter) (Table 1). These alterations had irreversible character persisted for the next 3 weeks after 4 weeks of L-NAME administration.
The results of the study indicate the presence of a pathogenetic link between endothelial dysfunction characterized by a lack of NO and hypertension. It was revealed that hypertension is associated with impaired endothelial function, and its correction (normalization of SP ad DP) is not always accompanied by normalization of endothelial function. Insufficient secretion of vasodilators (NO) causes an increase in the stiffness of the blood vessels’ walls and an increase in PP, which in turn contributes to the development of left ventricular hypertrophy and other serious complications in a living body. In response to the excess PP violation of the microcirculation of vital organs (such as the brain and kidneys), extensive tissue damage usually occurs [39]. Therefore, the therapeutic intervention aimed at increasing the bioavailability of NO may, therefore, be useful in conditions associated with age-related or premature arterial stiffening during hypertension.
Conclusions
The results of the study indicate the presence of a pathogenetic link between endothelial dysfunction and hypertension and the presence of residual endothelial dysfunction (characterized by insufficient NO) after the correction of hypertension. Therefore, in developing the new therapeutic approaches for the treatment of hypertension, it is necessary to include drugs that, in addition to correcting blood pressure, will support normalization, and potentiation of endothelial function and endogenous NO synthesis. However, the potential adverse effects of NO, including cytotoxicity, immune suppression and hypotensive shock, should also be taken into account.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
All subjects provided written informed consent.
Author Contributions
Irakli Megreladze and Nodar Mitagvaria designed and performed the study. Tamar Sanikidze drafted the manuscript and did critical editing. Ketavan Kakabadze and Nino Kvichia assisted and supported in sample collection and subsequent analysis with statistics. Nina Kipiani and Megi Dumbadze carefully supervised this manuscript preparation and writing.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Taddei S, Virdis A, Mattei P, Ghiadoni L, Sudano I, Salvetti A. Defective L-arginine-nitric oxide pathway in offspring of essential hypertensive patients. Circulation. 1996;94(6):1298-1303.
doi pubmed - Benjamin EJ, Larson MG, Keyes MJ, Mitchell GF, Vasan RS, Keaney JF, Jr., Lehman BT, et al. Clinical correlates and heritability of flow-mediated dilation in the community: the Framingham Heart Study. Circulation. 2004;109(5):613-619.
doi pubmed - Hermann M, Flammer A, Luscher TF. Nitric oxide in hypertension. J Clin Hypertens (Greenwich). 2006;8(12 Suppl 4):17-29.
doi pubmed - Vitecek J, Lojek A, Valacchi G, Kubala L. Arginine-based inhibitors of nitric oxide synthase: therapeutic potential and challenges. Mediators Inflamm. 2012;2012:318087.
doi pubmed - Kopincova J, Puzserova A, Bernatova I. L-NAME in the cardiovascular system - nitric oxide synthase activator? Pharmacol Rep. 2012;64(3):511-520.
doi - Southan GJ, Szabo C. Selective pharmacological inhibition of distinct nitric oxide synthase isoforms. Biochem Pharmacol. 1996;51(4):383-394.
doi - Wang YX, Poon CI, Pang CC. In vitro and ex vivo inhibitory effects of L- and D-enantiomers of NG-nitro-arginine on endothelium-dependent relaxation of rat aorta. J Pharmacol Exp Ther. 1993;265(1):112-119.
- Luszczki JJ, Jaskolska A, Dworzanski W, Zolkowska D. 7-Nitroindazole, but not NG-nitro-L-arginine, enhances the anticonvulsant activity of pregabalin in the mouse maximal electroshock-induced seizure model. Pharmacol Rep. 2011;63(1):169-175.
doi - Talarek S, Listos J, Fidecka S. Effect of nitric oxide synthase inhibitors on benzodiazepine withdrawal in mice and rats. Pharmacol Rep. 2011;63(3):680-689.
doi - Furfine ES, Harmon MF, Paith JE, Garvey EP. Selective inhibition of constitutive nitric oxide synthase by L-NG-nitroarginine. Biochemistry. 1993;32(33):8512-8517.
doi pubmed - Abdel-Rahman RF, Hessin AF, Abdelbaset M, Ogaly HA, Abd-Elsalam RM, Hassan SM. Antihypertensive effects of roselle-olive combination in L-NAME-induced hypertensive rats. Oxid Med Cell Longev. 2017;2017:9460653.
doi pubmed - Cebova M, Klimentova J, Janega P, Pechanova O. Effect of bioactive compound of Aronia melanocarpa on cardiovascular system in experimental hypertension. Oxid Med Cell Longev. 2017;2017:8156594.
doi pubmed - Miranda KM, Espey MG, Wink DA. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide. 2001;5(1):62-71.
doi pubmed - Massion PB, Feron O, Dessy C, Balligand JL. Nitric oxide and cardiac function: ten years after, and continuing. Circ Res. 2003;93(5):388-398.
doi pubmed - Vrankova S, Zemancikova A, Torok J, Pechanova O. Effect of low dose L-NAME pretreatment on nitric oxide/reactive oxygen species balance and vasoactivity in L-NAME/salt-induced hypertensive rats. J Physiol Pharmacol. 2019;70(4):535-544.
- Grumbach IM, Chen W, Mertens SA, Harrison DG. A negative feedback mechanism involving nitric oxide and nuclear factor kappa-B modulates endothelial nitric oxide synthase transcription. J Mol Cell Cardiol. 2005;39(4):595-603.
doi pubmed - Pechanova O, Simko F. Chronic antioxidant therapy fails to ameliorate hypertension: potential mechanisms behind. J Hypertens Suppl. 2009;27(6):S32-36.
doi pubmed - Zhen J, Lu H, Wang XQ, Vaziri ND, Zhou XJ. Upregulation of endothelial and inducible nitric oxide synthase expression by reactive oxygen species. Am J Hypertens. 2008;21(1):28-34.
doi pubmed - Sun Y, Carretero OA, Xu J, Rhaleb NE, Yang JJ, Pagano PJ, Yang XP. Deletion of inducible nitric oxide synthase provides cardioprotection in mice with 2-kidney, 1-clip hypertension. Hypertension. 2009;53(1):49-56.
doi pubmed - Xu H, Pritchard KA, Jr. Targeted increases in endothelial cell superoxide anion production stimulate eNOS-dependent nitric oxide production, not uncoupled eNOS activity. Arterioscler Thromb Vasc Biol. 2008;28(9):1580-1581.
doi pubmed - Carlstrom M, Liu M, Yang T, Zollbrecht C, Huang L, Peleli M, Borniquel S, et al. Cross-talk between Nitrate-Nitrite-NO and NO synthase pathways in control of vascular NO homeostasis. Antioxid Redox Signal. 2015;23(4):295-306.
doi pubmed - Vallance P, Collier J, Moncada S. Nitric oxide synthesised from L-arginine mediates endothelium dependent dilatation in human veins in vivo. Cardiovasc Res. 1989;23(12):1053-1057.
doi pubmed - Furchgott RF. Endothelium-dependent relaxation, endothelium-derived relaxing factor and photorelaxation of blood vessels. Semin Perinatol. 1991;15(1):11-15.
- Nava E, Palmer RMJ, Moncada S. FRS Inhibition of nitric oxide synthesis in septic shock: how much is beneficial? The Lancet. 1991;338(28):8782-8783.
doi - Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J. 1989;3(9):2007-2018.
doi - Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25(5):932-943.
doi pubmed - Pfeiffer S, Leopold E, Schmidt K, Brunner F, Mayer B. Inhibition of nitric oxide synthesis by NG-nitro-L-arginine methyl ester (L-NAME): requirement for bioactivation to the free acid, NG-nitro-L-arginine. Br J Pharmacol. 1996;118(6):1433-1440.
doi pubmed - Pechanova O, Bernatova I, Pelouch V, Babal P. L-NAME-induced protein remodeling and fibrosis in the rat heart. Physiol Res. 1999;48(5):353-362.
- Simko F, Simko J. The potential role of nitric oxide in the hypertrophic growth of the left ventricle. Physiol Res. 2000;49(1):37-46.
- Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, Geppetti P, et al. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest. 1994;94(5):2036-2044.
doi pubmed - Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100(12):3131-3139.
doi pubmed - Gerova M, Kristek F. Efficiency of NO donors in substituting impaired endogenous NO production: a functional and morphological study. Physiol Res. 2001;50(2):165-173.
- Simko F, Matuskova J, Luptak I, Pincikova T, Krajcirovicova K, Stvrtina S, Pomsar J, et al. Spironolactone differently influences remodeling of the left ventricle and aorta in L-NAME-induced hypertension. Physiol Res. 2007;56(Suppl 2):S25-32.
- Shirwany NA, Zou MH. Arterial stiffness: a brief review. Acta Pharmacol Sin. 2010;31(10):1267-1276.
doi pubmed - Fitch RM, Vergona R, Sullivan ME, Wang YX. Nitric oxide synthase inhibition increases aortic stiffness measured by pulse wave velocity in rats. Cardiovasc Res. 2001;51(2):351-358.
doi - Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J. 2010;4:302-312.
doi pubmed - Xu J, Shi GP. Vascular wall extracellular matrix proteins and vascular diseases. Biochim Biophys Acta. 2014;1842(11):2106-2119.
doi pubmed - Kanno Y, Into T, Lowenstein CJ, Matsushita K. Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovasc Res. 2008;77(1):221-230.
doi pubmed - Avolio AP, Kuznetsova T, Heyndrickx GR, Kerkhof PLM, Li JK-J. Arterial flow, pulse pressure, and pulse wave velocity in men and women at various ages. Adv Exp Med Biol. 2008;1065:53-168.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.