

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Review
Volume 15, Number 5, October 2024, pages 319-329

Diverse Concepts in Definitions of Dilated Cardiomyopathy: Theory and Practice
Michael C. Myersa, Boris Breznenb, Yue Zhonga, Sonomi Maruyamaa, Cindy Buenoa, Arnaud Bastiena, Mir Sohail Fazelib, Negar Golchina, c
aBristol Myers Squibb, Princeton, NJ, USA
bEvidinno Outcomes Research Inc., Vancouver, Canada
cCorresponding Author: Negar Golchin, Bristol Myers Squibb, Princeton, NJ, USA
Manuscript submitted July 5, 2024, accepted August 16, 2024, published online September 16, 2024
Short title: Definitions of Dilated Cardiomyopathy
doi: https://doi.org/10.14740/cr1679
| Abstract | ▴Top |
Our understanding of dilated cardiomyopathy (DCM) is evolving as new insights into the underlying pathophysiology become available. Professional organizations and clinical experts are improving definitions of DCM, allowing for more accurate treatment recommendations. This review summarized key published literature describing definitions and/or diagnostic criteria for DCM. Embase was searched from database inception to September 19, 2022 for 1) publications reporting definitions of DCM by major professional organizations and related opinion papers, and 2) clinical studies in DCM and heart failure with reduced ejection fraction. Sixty-eight records were included in this review. Definitions of DCM provided by two major professional organizations (American Heart Association (AHA) and European Society of Cardiology (ESC)) agreed on the clinical presentation of DCM; however, they differed in the classification of DCM within the larger context of cardiomyopathy taxonomies. Both organizations agreed that DCM could be clinically defined by the presence of left ventricular dilation and contractile dysfunction in the absence of abnormal loading conditions and severe coronary artery disease. AHA guidelines divided cardiomyopathies into two major groups (primary and secondary) based on predominant organ involvement. DCM was classified as primary cardiomyopathy with mixed (genetic and/or acquired) etiology. Conversely, ESC published a clinically oriented taxonomy in which cardiomyopathies were grouped into specific morphological and functional phenotypes; each was subclassified into familial or non-familial forms. Opinion papers further elaborated on the complex interplay between genetics and environment in the etiology of DCM. Several articles summarized the importance of the new and updated diagnostic tools, such as cardiac magnetic resonance imaging, electrocardiogram, and other biomarkers, in correctly identifying the etiology of DCM. Within clinical studies, most inclusion criteria used standard definitions proposed by leading professional associations (AHA and ESC). Clinical study investigators sometimes used a narrower definition of DCM using additional criteria for the left ventricular ejection fraction threshold value and left ventricular dilatation. Current efforts in cardiology research are focused on a more granular understanding of DCM etiology and the natural history of the disease. Definitions of DCM found in clinical studies mainly rely on published guidelines, with some studies adding idiosyncratic inclusion criteria refining the broad definitions of DCM.
Keywords: Dilated cardiomyopathy; Heart failure; Cardiology; Genetic dilated cardiomyopathy; Idiopathic dilated cardiomyopathy
| Introduction | ▴Top |
Cardiomyopathy is a myocardial disorder in which the heart muscle is structurally and functionally abnormal in the absence of coronary artery disease or abnormal loading conditions sufficient to cause the observed myocardial abnormality [1]. This abnormality can manifest as a mechanical problem such as systolic or diastolic dysfunction, or electrophysiological malfunction as seen in arrythmias. Cardiomyopathy is a generic phenotype that covers various pathophysiological conditions with clinical manifestations ranging from microscopic alterations in cardiac myocytes to fulminant heart failure (HF) with insufficient tissue perfusion, fluid accumulation, and cardiac arrythmia [2]. Finding the proper classification of the variety of subtypes that fall under the umbrella term of cardiomyopathy has been an ongoing effort of all major professional associations, as it is essential for diagnosing and treating the disease. This task has been complicated by overlapping symptomology of different heart conditions, which often leads to difficulties in identifying the correct underlying etiology [3-5]. In particular, the overlapping symptoms of HF and cardiomyopathy often present a challenge of correctly identifying the underlying disease and devising an appropriate treatment. Creating a classification scheme for cardiomyopathies can be approached from different angles. The traditional classification has been based on the clinical presentation of the ventricular morphology and function and it divided cardiomyopathies into hypertrophic, dilated, restrictive, and right ventricular arrhythmogenic cardiomyopathies [1, 6]. This classification has been recently updated to include our enhanced understanding of the underlying physiology, genetics, and natural history of the disease [7-9].
Among the subtypes of cardiomyopathy, dilated cardiomyopathy (DCM) is recognized as one of the most common causes of HF with reduced ejection fraction (HFrEF) and the leading indication for heart transplants worldwide [10]. The reported prevalence of DCM is around 36 cases per 100,000 individuals [9] and the annual incidence has been estimated as seven cases per 100,000 individuals [10]. In addition, DCM is the most common type of cardiomyopathy in children accounting for around 60% of all pediatric cardiomyopathies [10]. The broad definition of DCM is left or biventricular systolic dysfunction, often associated with dilation, in the absence of abnormal loading conditions or significant coronary artery disease [11]. However, this broad phenotype serves as a starting point for further refinement of the diagnosis by identifying the etiology, toxicology, and molecular and genetic factors of each patient. The ultimate goal is personalized treatment through precision medicine respecting the specifics of each case.
The challenge for the proper diagnostics of DCM in routine clinical practice is the evolving understanding of the disease and changing definitions and diagnostic criteria. Two major professional societies (American Heart Association (AHA) and European Society for Cardiology (ESC)) published recent updates of their classification schemes and definitions of DCM [7, 12, 13]. A comprehensive classification scheme for cardiomyopathies was also proposed by the World Heart Federation [14]. In addition, several high-profile position papers further refined our understanding of the variety of subtypes of DCM [11, 13, 15-17]. The evolving understanding of DCM reflected in the continuously updated DCM definitions creates a challenge for clinical practitioners to keep up with the current knowledge. This review is aimed at helping clinicians understand the existing definitions of DCM and its position within a larger typology of cardiomyopathies.
The objective of this literature review was to describe and characterize the landscape of evidence with respect to the definition and/or criteria used to diagnose DCM. Specifically, we aimed to identify the definitions of DCM (or similar diseases including non-ischemic cardiomyopathy and non-ischemic HFrEF) as reported by the most prominent and widely used clinical practice guidelines. Additionally, we aimed to summarize the status of adherence to these definitions in key clinical studies investigating DCM or HFrEF populations.
| Methods | ▴Top |
Standard methodologies for conducting and reporting systematic reviews recommended by the Cochrane Handbook for Systematic Reviews of Interventions [18], adapted for conducting a targeted literature review, were followed. Using predefined search strategies (Supplementary Material 1, www.cardiologyres.org), Embase was searched via the Ovid platform from database inception to September 19, 2022. Searches of bibliographies of included literature reviews were also conducted, as well as manual searches of Google Scholar to capture studies that were not included in the main literature database. Included were clinical practice guidelines, position papers, literature reviews, or clinical studies (observational studies or clinical trials) that described the definitions or other criteria used to diagnose DCM or similar diseases including non-ischemic cardiomyopathy and non-ischemic HFrEF. Articles were restricted to those published in English. Studies on perinatal cardiomyopathy or case reports/series were excluded.
All abstracts identified by the search were reviewed by a senior reviewer according to predefined eligibility criteria. All studies identified as eligible during the title and abstract screening were then screened at the full-text stage by the same reviewer. Reasons for inclusion or exclusion were documented, and relevant full-text articles identified at this stage were included for evidence synthesis. A Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) diagram was generated for complete transparency and reproducibility of the search and screening process [19].
The evidence base was split into two sets: publications reporting on definitions of DCM by major professional organizations and related opinion papers; and studies reporting on clinical studies in DCM and HFrEF. For the second set of studies, a standardized data extraction table was generated to define the study characteristics and outcomes that were extracted from eligible studies. Results were summarized in a narrative form highlighting the most relevant developments in the evolving definitions of DCM and their utilization in clinical studies.
| Results | ▴Top |
Study selection
A PRISMA flow diagram of the study selection procedure is presented in Figure 1. In total, 2,982 abstracts were identified from the literature searches including 2,976 records via Embase and six additional records through manual searches. The final set of included records consisted of 68 articles. Of the 68, seven were guidelines, 26 were position papers or narrative reviews, and one was a systematic literature review. These 34 articles were included in the synthesis of data to describe the definition and/or criteria used to diagnose DCM. The remaining 34 records were clinical studies that were used to summarize the status of adherence to these definitions and criteria.
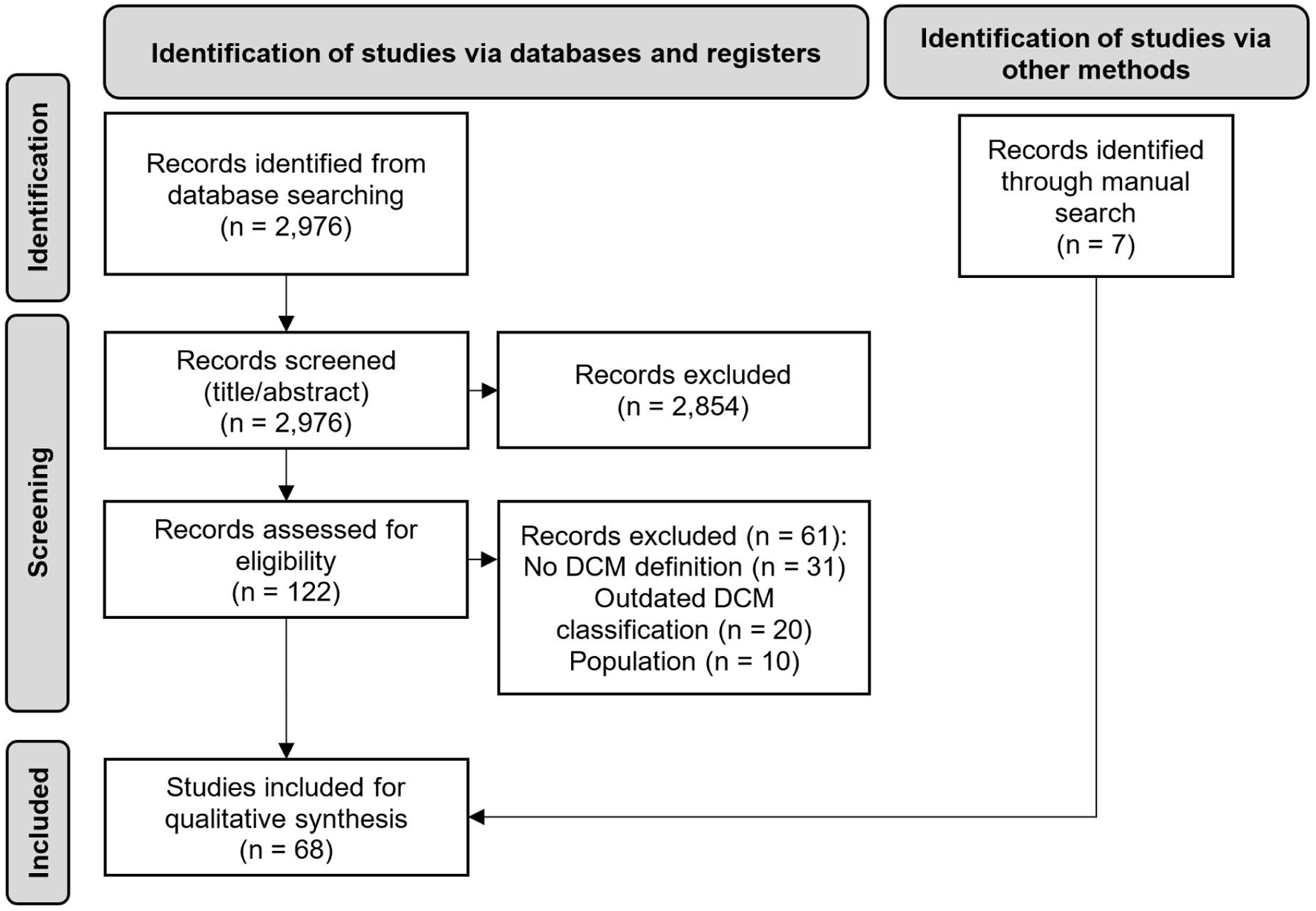 Click for large image | Figure 1. PRISMA diagram. n: number of records. |
Definitions of DCM
Professional societies’ guidelines
Currently, the most influential paper on the classification of cardiomyopathies was published in 2008 by the ESC Working Group on Myocardial and Pericardial Diseases [1]. The main motivation for developing this classification system was to provide a clinically oriented classification system in which heart muscle disorders are grouped according to ventricular morphology and function, rather than by etiology and genetics. Cardiomyopathies are grouped into specific morphological and functional phenotypes; each phenotype is then subclassified into familial and non-familial forms. In this context, “familial” refers to the occurrence in more than one family member of either the same disorder or a phenotype that is (or could be) caused by the same genetic mutation and not by acquired cardiac or systemic diseases in which the clinical phenotype is influenced by genetic polymorphism. Non-familial cardiomyopathies are clinically defined by the presence of cardiomyopathy in the index patient and the absence of disease in other family members (based on pedigree analysis and clinical evaluation). Non-familial cardiomyopathies are subdivided into idiopathic (no identifiable cause) and acquired (developed because of another disease, condition, or factor) cardiomyopathies in which ventricular dysfunction is a complication of the disorder, rather than an intrinsic feature of the disease [1].
The definition of DCM in this 2008 ESC guideline is as follows: “DCM is defined by the presence of left ventricular dilatation and left ventricular systolic dysfunction in the absence of abnormal loading conditions (hypertension (HTN), valve disease) or coronary artery disease sufficient to cause global systolic impairment. Right ventricular dilation and dysfunction may be present but are not necessary for the diagnosis” [1].
The same ESC Working Group that published the above guidelines further revised the definition of DCM in a separate document published in 2016 [13]. The aim of this position paper was to update the definition of DCM to take into account its diverse etiology and clinical manifestations in patients and relatives. The extended definition of DCM is as follows: “Left ventricular or biventricular systolic dysfunction and dilatation that are not explained by abnormal loading conditions or coronary artery disease” [13]. The authors provide specific criteria for systolic dysfunction and left ventricular dilation. An important update in the definition and understanding of DCM was the observation that the spectrum of electrical and functional abnormalities associated with this indication changes over time. This applies particularly to genetic diseases that have delayed or resulted in incomplete cardiac expression, with the result that many mutation carriers have intermediate phenotypes that do not meet standard disease definitions. For these reasons, the authors believe that clinical diagnosis and ultimately treatment can be improved by updating the criteria for diagnosis in relatives of DCM patients and the creation of a new category of hypokinetic non-dilated cardiomyopathy (HNDC). This transitory condition was defined as: “Left ventricular or biventricular global systolic dysfunction without dilatation (defined as LVEF [left ventricular ejection fraction] <45%), not explained by abnormal loading conditions or coronary artery disease” [13]. In the latest update to cardiomyopathy guidelines [12], ESC included a few novel phenotypic descriptions and simplified terminology used to describe the conditions. The five major phenotypes identified in this nomenclature are: hypertrophic cardiomyopathy; dilated cardiomyopathy; non-dilated left ventricular cardiomyopathy; arrhythmogenic right ventricular cardiomyopathy; and restrictive cardiomyopathy. The definition of DCM in this update remained the same as in the 2016 document [13]; however, the authors identified a new condition “isolated left ventricular dilatation” described as left ventricular dilatation that occurs with normal ejection fraction in the absence of athletic remodeling or other environmental factors. Authors noted that this is not in itself a cardiomyopathy but may represent an early manifestation of DCM [12].
The AHA approached the classification of cardiomyopathies from a different angle. The reference framework published by AHA in 2006 was based on a scientific presentation that offered new perspectives to aid in understanding this complex and heterogeneous group of diseases and basic disease mechanisms [6]. The classification of cardiomyopathies presented in this document was not intended to provide precise methodologies or strategies for clinical diagnosis, but rather it relied on contemporary molecular biology, considering cellular levels of expression of encoded proteins and underlying gene mutations. Consequently, cardiomyopathies are divided into two major groups based on predominant organ involvement. Primary cardiomyopathies (genetic, non-genetic, and acquired) are those solely or predominantly confined to the heart muscle and are relatively few. Secondary cardiomyopathies show pathological myocardial involvement as part of a large number and variety of generalized systemic (multiorgan) disorders. In this scheme, DCM is classified under the “mixed” label, meaning that it can fall under both genetic and acquired etiology. There is no explicit definition of DCM in this framework, only a clinical description of DCM characteristics: “Dilated forms of cardiomyopathy are characterized by ventricular chamber enlargement and systolic dysfunction with normal [left ventricular] wall thickness; usually diagnosis is made with 2-dimensional echocardiography” [6]. A side-by-side comparison of the AHA and ESC classification schemes is shown in Table 1.
 Click to view | Table 1. Classification Schemes of Cardiomyopathies From AHA and ESC |
AHA released an updated scientific statement on diagnostics and treatment strategies for DCM in 2016 [7]. The definition of DCM was introduced as follows: “The term dilated cardiomyopathy (DCM) refers to a spectrum of heterogeneous myocardial disorders that are characterized by ventricular dilation and depressed myocardial performance in the absence of hypertension, valvular, congenital, or ischemic heart disease” [7]. The diagnostic and treatment recommendations for DCM subtypes are introduced for cardiac amyloidosis, cardiotoxins, peripartum cardiomyopathy, cardiac sarcoidosis, myocarditis, autoimmune cardiomyopathy, endocrine and metabolic cardiomyopathies, and genetic cardiomyopathies.
The World Heart Federation introduced a radically new classification of cardiomyopathies in 2013 for which the proposed classification is based on the phenotype-genotype distinction with respect to the pathology [14]. The authors propose a nosology that addresses five simple attributes of a cardiomyopathic disorder, including morpho-functional characteristic (M), organ involvement (O), genetic or familial inheritance pattern (G), and an explicit etiological annotation (E) with details of genetic defect or underlying disease/cause; information about the functional status (S) using the American College of Cardiology/AHA (A to D) stage of HF and New York Heart Association (NYHA, I to IV) functional classes of HF may also be added. The addition of (S) has been left optional and should be used at the discretion of the physician. With the description of five attributes, the classification system is designated as MOGE(S). In this classification scheme, each patient will be diagnosed by assigning specific values to each of the attributes mentioned above. For example, a patient with hypertrophic cardiomyopathy affecting the heart only, with autosomal dominant genetic inheritance, and genetic etiology involving the MYH7[p.R663h] gene would be assigned designation MHOHGADEG-MYH7[p.R663H].
Opinion papers
Merlo et al (2018) published an influential opinion paper on the evolving concepts in DCM [17]. The main issues addressed by this paper can be summarized into three categories. Category 1) contains the etiological characterization and early diagnosis of DCM. Given that DCM is characterized by complex interactions between environment and genetic predisposition, identifying all contributing factors early in the diagnosis is crucial for effective treatment. Examples of commonly overlooked or underappreciated reversible triggers for left ventricular dysfunction include certain types of arrythmias, substance abuse, acute emotional stress, chemotherapies, and systemic autoimmune diseases. Category 2) describes the interaction between genotype and clinical phenotype in DCM. So far, more than 50 genes encoding for sarcomeric proteins, cytoskeleton, nuclear envelope, sarcolemma, ion channels and intercellular junctions have been implicated in DCM. With a family history of DCM, genetic origin of the disease becomes likely; however, a negative family history does not rule out a genetic form of DCM as de novo mutations can be responsible for sporadic forms. Category 3) describes that DCM is a dynamic disease. In recent years, several studies revealed that almost 40% of patients experience significant left ventricular reverse remodeling when treated with evidence-based pharmacological and device treatments. This issue emphasizes the pivotal role not only of an accurate and complete initial diagnostic evaluation but also of continuous therapy and individualized, long-term surveillance to recognize and treat the first signs of a decline in systolic function.
Several papers surveyed the diagnostics challenges in the early detection of DCM and the differentiation from other cardiomyopathies [5, 20-22]. Moeinafshar et al (2021) provided a summary of the most relevant biomarkers of DCM [23]. Three categories of biomarkers identified in this review were cardiomyocyte-related, microenvironmental, and macroenvironmental. The importance of the respective biomarkers is related to the individual patient’s history and status at admission. Different biomarkers are relevant to, for example, hemodynamic function, inflammation, cardiac repair, and myocyte necrosis. Since a rapid and accurate diagnosis of cardiomyopathies is crucial in preventing HF and/or death, the novel biomarkers can play an increasingly important role as our understanding of their validity improves.
Before diagnosing DCM, it is necessary to exclude conditions with phenotypic overlap. Advanced diagnostic modalities are also increasingly important for the identification of the underlying etiology. The role of cardiac magnetic resonance imaging (MRI) in visualizing the morphology of the heart was reviewed by Aggarwal et al (2014) [4]. The authors describe the most relevant MRI pulse sequences and the diagnostics information they provide as well as typical MRI signatures corresponding to different cardiomyopathies. Finocchiaro et al (2020) summarized the recent advances in electrocardiogram (ECG) methodology for the diagnosis and risk stratification of DCM [24]. The traditional notion that ECG abnormalities in DCM are non-specific was challenged in studies that identified several “red flag” features associated with the genetic forms of DCM as well as ECG features found in the non-genetic forms of the disease. Heart rate variability as a potential biomarker for differentiation between early-stage ischemic heart disease and DCM was investigated by Accardo et al (2022) [3]. The proposed model based on pNN50 feature, fractal dimension, sex, age, and LVEF features achieved 73.3% accuracy in the differential diagnosis of ischemic heart disease vs. DCM.
HF and DCM
DCM is one of the leading causes of HF with its prevalence in the population with HF estimated to be between 8% and 47% [9]. Patients are also at risk of sudden cardiac death [25] and consequently DCM is also a common indication for implantable cardioverter defibrillators and heart transplantation [2]. Given the high incidence of DCM in HF, particularly in HFrEF, distinguishing DCM from other HF etiologies is critical for devising the proper treatment regimen. The 2016 ESC guidelines define HF as “… clinical syndrome characterized by typical symptoms (e.g., breathlessness, ankle swelling and fatigue) that may be accompanied by signs (e.g., elevated jugular venous pressure, pulmonary crackles and peripheral oedema) caused by a structural and/or functional cardiac abnormality, resulting in a reduced cardiac output and/or elevated intracardiac pressures at rest or during stress” [8]. DCM should be considered as a possible cause particularly in new-onset (“de novo”) HF where a patient can be presenting with symptoms for weeks or months preceding the HF diagnosis [8]. The natural history of HF in DCM can be characterized by three distinct pathways including: 1) a structural and functional recovery following incident HF; 2) remission of HF symptoms and improvement/stabilization of left ventricular systolic function; and 3) progression to advanced HF and heart transplantation/death. Each of these pathways requires a distinct diagnostic and therapeutic approach, therefore prior conceptual clarity is vital for successful intervention.
DCM in clinical studies
To investigate the adherence to the DCM definitions outlined in the guidance documents in clinical studies, 34 clinical studies (28 observational studies, three clinical trials, and three post-hoc analyses) were included in the evidence base [26-59]. The selection of the studies for this objective was based on the inclusion criteria of the studies as well as on the primary objectives described in the study. Studies investigating the DCM population or those investigating HF outcomes were included.
The population size in the included studies ranged from a minimum of 10 [39] (a retrospective analysis of 10 pediatric patients diagnosed with hypocalcemic DCM) to a maximum of 8,399 [32] (a post-hoc analysis of PARADIGM-HF clinical trial data). The mean population size was 548 and the median was 151.
Patient inclusion criteria in the included studies can be summarized into the following categories: 1) Adult patients with some form of DCM in 21 studies [29, 30, 33-38, 42-46, 48, 50, 51, 53, 55, 56, 58, 59]; 2) Pediatric patients with DCM in three studies [39, 40, 54]; 3) Patients with HF in four studies [26, 31, 32, 57]; and 4) Other inclusion criteria in six studies [27, 28, 41, 49, 52, 57]. In this category, the inclusion criteria usually required LVEF below a certain threshold, e.g., LVEF < 50% [28], LVEF < 40% [49, 57], or LVEF < 36% [52].
Only 14 of the 34 studies specified exclusion criteria. When provided, the exclusion criteria usually mentioned coronary artery disease, HTN, or other unrelated cardiomyopathies.
The included studies investigated a variety of endpoints: 1) Diagnosis of DCM in 14 studies [26-30, 33, 34, 36, 39-42, 55, 57]; 2) Prognosis of DCM patients in 10 studies [31, 37, 45, 46, 49, 51, 52, 54, 58, 59]; 3) Cardiac outcomes (including HF, cardiac death, major adverse cardiovascular events, relapse, and others) in seven studies [32, 35, 38, 43, 44, 48, 57]; and 4) Other outcomes (fibrosis-related outcomes, custom index evaluation) in three studies [50, 53, 56].
The definition of DCM in the included studies was mostly based on the standard definition of DCM as provided by the guidelines and position papers. The most often cited document was the position statement from the ESC published by Elliott et al (2008) [1]. Twelve studies used this reference for the definition of DCM. Two studies used the scientific statement from AHA published by Bozkurt et al (2016) [7]. Three studies used a position paper on the evolving concepts in DCM published by Merlo et al (2018) [17]. Two studies used an opinion paper published by Japp et al (2016) [60]. Two studies used the updated proposal for a revised definition of DCM published by Pinto et al (2016) [13].
Examples of typical definitions of DCM using the guidance documents are provided in Table 2 [34, 36, 37, 42, 45, 46, 49].
Click to view | Table 2. Example Definitions of DCM in Clinical Studies |
Some studies used additional criteria for defining the DCM. Those criteria were ubiquitous for each study and not seen in other studies. Examples of the most relevant studies using their own DCM criteria are below. 1) Akinrinade et al (2015) [30] published a study using a high-quality oligonucleotide-selective sequencing-based targeted sequencing panel to investigate the genetic landscape of DCM in the Finnish population and to evaluate the utility of oligonucleotide-selective sequencing technology as a novel comprehensive diagnostic tool. DCM was diagnosed using the following criteria: “… left ventricular (LV) end-diastolic diameter (LVEDD) 0.27 mm/m2 [modified from original criteria of 0.117% of the predicted value corrected for age and body surface area (BSA)] and left ventricular systolic dysfunction (LVEF < 45%) in the absence of abnormal loading conditions such as hypertensive heart disease, primary valve disease, or significant coronary artery disease.” 2) Balmforth et al (2019) published a post-hoc analysis of the PARADIGM-HF clinical trial data examining the outcomes and the effect of sacubitril/valsartan according to investigator-reported etiology [32]. The etiology of HF was collected by means of structured questions on the trial case report form. Investigators were first asked whether the primary etiology was ischemic or non-ischemic. If the answer non-ischemic was checked, investigators were then asked to specify from a number of options (listed in the following order): primary valvular (specify valve and surgery), alcoholic, hypertensive, idiopathic, peripartum, infectious cardiomyopathy, viral cardiomyopathy, diabetic, drug-induced (specify type of drug), and “other” (please specify). For the purposes of this analysis, patients were categorized as ischemic or non-ischemic, with non-ischemic etiology being further subcategorized into idiopathic, hypertensive and other, because the numbers of cases in “other” were individually too few to allow robust analysis.
| Discussion | ▴Top |
This review aimed to summarize the current understanding of DCM as reflected in guidance documents, recommendations, and position papers authored by the main professional associations and key opinion leaders. To understand the evolution of the current definitions of DCM, it is important to start from the overall classification of cardiomyopathies, as DCM is a subcategory of the broad family of diseases of the heart. Cardiomyopathies are a heterogeneous group of pathologies characterized by structural and functional alterations of the heart muscle. The two main associations of cardiac professionals - the AHA and the ESC - recently released their updated classification scheme for cardiomyopathies. The two classifications differ in their approach and consequently the resulting categories do not correspond directly to each other.
The ESC classification of cardiomyopathies was mostly motivated by the clinical relevance of the categories, and thus, in this scheme, the heart muscle disorders are grouped according to ventricular morphology and function [1]. Each phenotype is then subclassified into familial and non-familial forms. As a phenotype within this classification, DCM can present in both familial and non-familial forms and the identification of the correct etiology is essential for the correct diagnosis.
The definition of DCM was further revised by the ESC Working Group on Myocardial and Pericardial Diseases in 2016 with the aim of considering its diverse etiology and clinical manifestations in patients and relatives [13]. The definition of DCM was augmented by additional diagnostic criteria for left ventricular dilation, and the natural history of DCM was extended to the pre-clinical phase. The DCM clinical spectrum therefore starts with a potential mutation carrier with no cardiac expression and goes through progressive expression of the phenotype, with the first clinical phase defined as HNDC (a new category proposed in this 2016 ESC Working Group paper and reflected in 2021 ESC HF guidelines [61]) all the way to full-blown DCM. The emphasis in this document is on clinical diagnostics and differentiation of the DCM phenotype from other cardiomyopathies.
In contrast to ESC, the AHA classification of cardiomyopathies takes a broader and more scientific approach that incorporates the rapid evolution of molecular genetics in cardiology [6]. In a departure from the ESC approach, AHA noted that “functional (i.e., physiological) classifications, seemingly most useful to clinicians with relevance to treatment considerations, are in fact of limited value because management strategies are dynamic and inevitably evolve for these diseases” [6]. The resulting scheme proposed by AHA divides cardiomyopathies into two major groups based on predominant organ involvement: primary and secondary.
The classification hierarchies developed by the two professional organizations aim to guide appropriate diagnostic and treatment strategies that prevent the development and progression of HF in patients with specific cardiomyopathies [7]. The variety of causes, multiple underlying pathophysiological mechanisms, and different phenotypic expressions in cardiomyopathies influence their presentation and response to treatment [9]. In order to correctly identify the phenotype, the focus should be on proper diagnostics using our most recent understanding of the etiology and natural history of the disease. After establishing the phenotype, the guidelines offer treatment recommendations most suitable for each subtype of cardiomyopathy.
In addition to the professional organizations, several high-profile opinion papers have been published. The papers further elaborate on the complex interplay between genetics and environment in the etiology of DCM [17]. Several papers summarized the importance of the new and updated diagnostics tools such as cardiac MRI [4] and ECG [24] as well as other biomarkers [3, 23] in correctly identifying the etiology of DCM.
Since DCM is one of the leading causes of HF, guidance documents on HF also include considerations of the underlying etiology of HF [8, 9], which may include DCM. The documents note that in DCM, HFrEF has a high incidence and prevalence and represents the most frequent cause of death, despite improvements in treatment. In addition, advanced HF in DCM is one of the leading indications for heart transplantation. The definition of DCM in these documents broadly agrees with the standard definition proposed by ESC.
When analyzing clinical studies in DCM and HF, most of the inclusion criteria use the standard definition proposed by either of the two main professional associations (AHA and ESC). The most popular reference cited in clinical studies is the ESC classification of cardiomyopathies published by Elliott et al (2008) [1]. Other documents by ESC and AHA are cited as well. In a few cases, the researchers running the clinical study use a narrower definition of DCM using additional criteria for LVEF threshold value. The definition of DCM in the studies was usually pragmatic and used for selecting the patient population. The baseline definition was then used as a starting point for identifying prognostic biomarkers and risk factors of HF. The research focus was on finding physiological biomarkers for differential diagnosis of DCM versus other similar cardiomyopathies, biomarkers identifying high-risk patients, genetic markers associated with familial DCM, and biomarkers indicating reverse ventricular remodeling. The included studies did not feature recent updates in the definitions of DCM reflected in the guidelines; however, the research directions in the studies align with the overall advancements in the understanding of DCM typology outlined in the updated guidelines.
The targeted approach to conducting this review was the most appropriate methodology for answering the research questions at hand. The documents featured here are the most relevant and they were selected based on the affiliation of the authors and the authority of the professional organizations in the field of cardiology. The selection is therefore limited to directly relevant documents and does not include all opinion papers related to DCM. Many interesting papers explored issues of detailed etiology of DCM and advanced diagnostics techniques (such as artificial intelligence) used to further subdivide DCM into subcategories. As interesting and potentially important as those papers are, the topics of advanced diagnostics are outside the scope of this review.
The sample clinical trials were selected based on their inclusion criteria rather than their interventions and/or outcomes. The focus was on key published studies relevant to our research question, and therefore the sample may not be comprehensive of all studies enrolling DCM patients. The studies were selected to be representative of the current understanding of definitions of DCM within the context of its diagnostics and treatment. The success or failure of the interventions was not the primary concern.
| Conclusions | ▴Top |
DCM is a clinically heterogeneous disease with large variability in age of disease onset and rate of progression, which is likely explained by the complex interplay between genetic susceptibility and environmental factors. The definitions of DCM provided by the two major professional organizations agree about the clinical presentation of DCM but differ in the classification of DCM within the larger context of cardiomyopathy taxonomies. Both AHA and ESC agree that the diagnosis of DCM requires a more granular approach to its natural history and prognosis. Currently, most effort is focused on further differentiating the etiology of DCM based on the genetic profiling and environmental causes specific to each patient. To this end, new diagnostic tools are being investigated both in advanced genotyping as well as in imaging and in electrocardiography. The review of relevant clinical studies showed that most studies use a standardized definition of DCM as put forward by professional organizations with some modifications to the threshold values of LVEF. In conclusion, the umbrella definition of DCM is well accepted and widely used, but the main effort is now in refining the identification of subtypes of DCM and the natural progression of the disease with the goal of developing personalized treatment tailored for each patient.
| Supplementary Material | ▴Top |
Suppl 1. Search strategy for Embase (via OvidSP).
Acknowledgments
The authors would like to thank Kimberly Hofer and Masoud Pourrahmat of Evidinno Outcomes Research Inc. (Vancouver, BC) for their help with conducting the research.
Financial Disclosure
This study was funded by Bristol Myers Squibb.
Conflict of Interest
MCM, YZ, SM, CB, AB, and NG are employees and/or shareholders of Bristol Myers Squibb. BB and MSF are employed by Evidinno Outcomes Research Inc. (Vancouver, BC, Canada), which was contracted by Bristol Myers Squibb to conduct this study.
Author Contributions
Michael C. Myers: conceptualization, writing - review and editing, visualization, and supervision; Boris Breznen: investigation, validation, writing - original draft, writing - review and editing, visualization, and supervision; Yue Zhong: writing - review and editing, visualization, and supervision; Sonomi Maruyama: writing - review and editing, visualization, and supervision; Cindy Bueno: writing - review and editing, visualization, and supervision; Arnaud Bastien: writing - review and editing, visualization, and supervision; Mir Sohail Fazeli: conceptualization, methodology, writing - review and editing, visualization, and supervision; Negar Golchin: conceptualization, writing - review and editing, visualization, and supervision.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
AHA: American Heart Association; DCM: dilated cardiomyopathy; ECG: electrocardiogram; ESC: European Society of Cardiology; HF: heart failure; HFrEF: heart failure with reduced ejection fraction; HNDC: hypokinetic non-dilated cardiomyopathy; HT: hypertension; LV: left ventricular; LVEDD: left ventricular end-diastolic diameter; LVEF: left ventricular ejection fraction; MOGE(S): morpho-functional characteristic, organ involvement (O), genetic or familial inheritance pattern (G), and an explicit etiological annotation (E) with details of genetic defect or underlying disease/cause; information about the functional status (S): ; MRI: magnetic resonance imaging; NICM: non-ischemic cardiomyopathy; NYHA: New York Heart Association; PRISMA: Preferred Reporting Items for Systematic Reviews and Meta-Analyses
| References | ▴Top |
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270-276.
doi pubmed - Brieler J, Breeden MA, Tucker J. Cardiomyopathy: an overview. Am Fam Physician. 2017;96(10):640-646.
pubmed - Accardo A, Restivo L, Ajcevic M, Miladinovic A, Iscra K, Silveri G, Merlo M, et al. Toward a diagnostic CART model for Ischemic heart disease and idiopathic dilated cardiomyopathy based on heart rate total variability. Med Biol Eng Comput. 2022;60(9):2655-2663.
doi pubmed pmc - Aggarwal NR, Peterson TJ, Young PM, Araoz PA, Glockner J, Mankad SV, Williamson EE. Unveiling nonischemic cardiomyopathies with cardiac magnetic resonance. Expert Rev Cardiovasc Ther. 2014;12(2):217-239.
doi pubmed - Alimadadi A, Manandhar I, Aryal S, Munroe PB, Joe B, Cheng X. Machine learning-based classification and diagnosis of clinical cardiomyopathies. Physiol Genomics. 2020;52(9):391-400.
doi pubmed pmc - Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807-1816.
doi pubmed - Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, Fonarow GC, Francis GS, et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation. 2016;134(23):e579-e646.
doi pubmed - Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2016;18(8):891-975.
doi pubmed - Seferovic PM, Polovina M, Bauersachs J, Arad M, Ben Gal T, Lund LH, Felix SB, et al. Heart failure in cardiomyopathies: a position paper from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2019;21(5):553-576.
doi pubmed - Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. 2017;390(10092):400-414.
doi pubmed - Manca P, Nuzzi V, Cannata A, Merlo M, Sinagra G. Contemporary etiology and prognosis of dilated non-ischemic cardiomyopathy. Minerva Cardiol Angiol. 2022;70(2):171-188.
doi pubmed - Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C, Bezzina CR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44(37):3503-3626.
doi pubmed - Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Bohm M, Duboc D, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37(23):1850-1858.
doi pubmed - Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the world heart federation. Glob Heart. 2013;8(4):355-382.
doi pubmed - Bakalakos A, Ritsatos K, Anastasakis A. Current perspectives on the diagnosis and management of dilated cardiomyopathy Beyond heart failure: a Cardiomyopathy Clinic Doctor's point of view. Hellenic J Cardiol. 2018;59(5):254-261.
doi pubmed - Laurence C, Fenton M. Dilated cardiomyopathy: room for (cautious) optimism? J Paediatr Child Health. 2021;31(2):68-74.
- Merlo M, Cannata A, Gobbo M, Stolfo D, Elliott PM, Sinagra G. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail. 2018;20(2):228-239.
doi pubmed - Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA. Cochrane handbook for systematic reviews of interventions. 2nd ed: John Wiley & Sons; 2019.
- Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, Shamseer L, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.
doi pubmed pmc - Palmiero G, Carlomagno G, Lucivero G. Diagnosis of cardiomyopathies: Tips and tricks for internists and general practitioners. Cardiogenetics. 2017;7(1):6-17
- Sammani A, Baas AF, Asselbergs FW, Te Riele A. Diagnosis and risk prediction of dilated cardiomyopathy in the era of big data and genomics. J Clin Med. 2021;10(5):921.
doi pubmed pmc - Tayal U, Verdonschot JAJ, Hazebroek MR, Howard J, Gregson J, Newsome S, Gulati A, et al. Precision phenotyping of dilated cardiomyopathy using multidimensional data. J Am Coll Cardiol. 2022;79(22):2219-2232.
doi pubmed pmc - Moeinafshar A, Yazdanpanah N, Rezaei N. Diagnostic biomarkers of dilated cardiomyopathy. Immunobiology. 2021;226(6):152153.
doi pubmed - Finocchiaro G, Merlo M, Sheikh N, De Angelis G, Papadakis M, Olivotto I, Rapezzi C, et al. The electrocardiogram in the diagnosis and management of patients with dilated cardiomyopathy. Eur J Heart Fail. 2020;22(7):1097-1107.
doi pubmed - Halliday BP, Cleland JGF, Goldberger JJ, Prasad SK. Personalizing risk stratification for sudden death in dilated cardiomyopathy: the past, present, and future. Circulation. 2017;136(2):215-231.
doi pubmed pmc - Abunassar JG, Yam Y, Chen L, D'Mello N, Chow BJ. Usefulness of the Agatston score = 0 to exclude ischemic cardiomyopathy in patients with heart failure. Am J Cardiol. 2011;107(3):428-432.
doi pubmed - Adam M, Oh SL, Sudarshan VK, Koh JE, Hagiwara Y, Tan JH, Tan RS, et al. Automated characterization of cardiovascular diseases using relative wavelet nonlinear features extracted from ECG signals. Comput Methods Programs Biomed. 2018;161:133-143.
doi pubmed - Aghasadeghi K, Aslani A. Differentiation of ischemic and dilated cardiomyopathy on electrocardiograms. Asian Cardiovasc Thorac Ann. 2008;16(2):103-106.
doi pubmed - Akinrinade O, Helio T, Lekanne Deprez RH, Jongbloed JDH, Boven LG, van den Berg MP, Pinto YM, et al. Relevance of titin missense and non-frameshifting insertions/deletions variants in dilated cardiomyopathy. Sci Rep. 2019;9(1):4093.
doi pubmed pmc - Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenpera P, Koillinen H, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36(34):2327-2337.
doi pubmed pmc - Alla F, Briancon S, Juilliere Y, Mertes PM, Villemot JP, Zannad F. Differential clinical prognostic classifications in dilated and ischemic advanced heart failure: the EPICAL study. Am Heart J. 2000;139(5):895-904.
doi pubmed - Balmforth C, Simpson J, Shen L, Jhund PS, Lefkowitz M, Rizkala AR, Rouleau JL, et al. Outcomes and effect of treatment according to etiology in HFrEF: an analysis of PARADIGM-HF. JACC Heart Fail. 2019;7(6):457-465.
doi pubmed - Behera BK, Radha Krishna Naik M, Jali SN, Tripathy S, Behera N. A study of clinical and echocardiographic profile in patients of dilated cardiomyopathy. Asian J Pharm Clin Res. 2022;15(8):144-150.
- Calderon-Dominguez M, Belmonte T, Quezada-Feijoo M, Ramos M, Calderon-Dominguez J, Campuzano O, Mangas A, et al. Plasma microrna expression profile for reduced ejection fraction in dilated cardiomyopathy. Sci Rep. 2021;11(1):7517.
doi pubmed pmc - Canu M, Margerit L, Mekhdoul I, Broisat A, Riou L, Djaileb L, Charlon C, et al. Prognosis of coronary atherosclerotic burden in non-ischemic dilated cardiomyopathies. J Clin Med. 2021;10(10):2183.
doi pubmed pmc - Costa MC, Calderon-Dominguez M, Mangas A, Campuzano O, Sarquella-Brugada G, Ramos M, Quezada-Feijoo M, et al. Circulating circRNA as biomarkers for dilated cardiomyopathy etiology. J Mol Med (Berl). 2021;99(12):1711-1725.
doi pubmed pmc - Diez-Lopez C, Salazar-Mendiguchia J, Garcia-Romero E, Fuentes L, Lupon J, Bayes-Genis A, Manito N, et al. Clinical determinants and prognosis of left ventricular reverse remodelling in non-ischemic dilated cardiomyopathy. J Cardiovasc Dev Dis. 2022;9(1):20.
doi pubmed pmc - Feng J, Tian P, Liang L, Chen Y, Wang Y, Zhai M, Huang Y, et al. Outcome and prognostic value of N-terminal pro-brain natriuretic peptide and high-sensitivity C-reactive protein in mildly dilated cardiomyopathy vs. dilated cardiomyopathy. ESC Heart Fail. 2022;9(3):1625-1635.
doi pubmed pmc - Garg A, Azad S, Kumar K, Bhatia M, Radhakrishnan S. Role of cardiac magnetic resonance imaging in hypocalcemia-induced dilated cardiomyopathy in pediatric population. Indian J Radiol Imaging. 2021;31(4):837-843.
doi pubmed pmc - Gran F, Fidalgo A, Dolader P, Garrido M, Navarro A, Izquierdo-Blasco J, Balcells J, et al. Differences between genetic dilated cardiomyopathy and myocarditis in children presenting with severe cardiac dysfunction. Eur J Pediatr. 2022;181(1):287-294.
doi pubmed pmc - Guelly C, Abilova Z, Nuralinov O, Panzitt K, Akhmetova A, Rakhimova S, Kozhamkulov U, et al. Patients with coronary heart disease, dilated cardiomyopathy and idiopathic ventricular tachycardia share overlapping patterns of pathogenic variation in cardiac risk genes. PeerJ. 2021;9:e10711.
doi pubmed pmc - Haas GJ, Zareba KM, Ni H, Bello-Pardo E, Huggins GS, Hershberger RE, Study Principal I, et al. Validating an idiopathic dilated cardiomyopathy diagnosis using cardiovascular magnetic resonance: the dilated cardiomyopathy precision medicine study. Circ Heart Fail. 2022;15(5):e008877.
doi pubmed pmc - Halliday BP, Owen R, Gregson J, V SV, Chen X, Wage R, Lota AS, et al. Myocardial remodelling after withdrawing therapy for heart failure in patients with recovered dilated cardiomyopathy: insights from TRED-HF. Eur J Heart Fail. 2021;23(2):293-301.
doi pubmed - Halliday BP, Wassall R, Lota AS, Khalique Z, Gregson J, Newsome S, Jackson R, et al. Withdrawal of pharmacological treatment for heart failure in patients with recovered dilated cardiomyopathy (TRED-HF): an open-label, pilot, randomised trial. Lancet. 2019;393(10166):61-73.
doi pubmed pmc - Hamshere S, Arnous S, Choudhury T, Choudry F, Mozid A, Yeo C, Barrett C, et al. Randomized trial of combination cytokine and adult autologous bone marrow progenitor cell administration in patients with non-ischaemic dilated cardiomyopathy: the REGENERATE-DCM clinical trial. Eur Heart J. 2015;36(44):3061-3069.
doi pubmed pmc - Kimura Y, Okumura T, Morimoto R, Kazama S, Shibata N, Oishi H, Araki T, et al. A clinical score for predicting left ventricular reverse remodelling in patients with dilated cardiomyopathy. ESC Heart Fail. 2021;8(2):1359-1368.
doi pubmed pmc - Manca P, Stolfo D, Merlo M, Gregorio C, Cannata A, Ramani F, Nuzzi V, et al. Transient versus persistent improved ejection fraction in non-ischaemic dilated cardiomyopathy. Eur J Heart Fail. 2022;24(7):1171-1179.
doi pubmed - Martino HF, Oliveira PS, Souza FC, Costa PC, Assuncao ESE, Villela R, Gaze M, et al. A safety and feasibility study of cell therapy in dilated cardiomyopathy. Braz J Med Biol Res. 2010;43(10):989-995.
doi pubmed - Merlo M, Mase M, Perry A, La Franca E, Deych E, Ajello L, Bellavia D, et al. Prognostic significance of longitudinal strain in dilated cardiomyopathy with recovered ejection fraction. Heart. 2022;108(9):710-716.
doi pubmed pmc - Mikami Y, Cornhill A, Dykstra S, Satriano A, Hansen R, Flewitt J, Seib M, et al. Right ventricular insertion site fibrosis in a dilated cardiomyopathy referral population: phenotypic associations and value for the prediction of heart failure admission or death. J Cardiovasc Magn Reson. 2021;23(1):79.
doi pubmed pmc - Ohta-Ogo K, Sugano Y, Ogata S, Nakayama T, Komori T, Eguchi K, Dohi K, et al. Myocardial T-lymphocytes as a prognostic risk-stratifying marker of dilated cardiomyopathy- results of the multicenter registry to investigate inflammatory cell infiltration in dilated cardiomyopathy in tissues of endomyocardial biopsy (INDICATE Study). Circ J. 2022;86(7):1092-1101.
doi pubmed - Rashba EJ, Estes NA, Wang P, Schaechter A, Howard A, Zareba W, Couderc JP, et al. Preserved heart rate variability identifies low-risk patients with nonischemic dilated cardiomyopathy: results from the DEFINITE trial. Heart Rhythm. 2006;3(3):281-286.
doi pubmed - Rubis PP, Dziewiecka EM, Banys P, Urbanczyk-Zawadzka M, Krupinski M, Mielnik M, Lach J, et al. Extracellular volume is an independent predictor of arrhythmic burden in dilated cardiomyopathy. Sci Rep. 2021;11(1):24000.
doi pubmed pmc - Seidel F, Holtgrewe M, Al-Wakeel-Marquard N, Opgen-Rhein B, Dartsch J, Herbst C, Beule D, et al. Pathogenic variants associated with dilated cardiomyopathy predict outcome in pediatric myocarditis. Circ Genom Precis Med. 2021;14(4):e003250.
doi pubmed pmc - Stroeks S, Hellebrekers D, Claes GRF, Tayal U, Krapels IPC, Vanhoutte EK, van den Wijngaard A, et al. Clinical impact of re-evaluating genes and variants implicated in dilated cardiomyopathy. Genet Med. 2021;23(11):2186-2193.
doi pubmed pmc - Valverde-Gomez M, Ruiz-Curiel A, Melendo-Viu M, Salguero-Bodes R, Martin-Arriscado C, Bueno H, Jimenez-Lopez-Guarch C, et al. Electrocardiogram changes in the spectrum of TTNtv dilated cardiomyopathy: accuracy and predictive value of a new index for LV-changes identification. Heart Lung Circ. 2021;30(10):1487-1495.
doi pubmed - Vera A, Cecconi A, Martinez-Vives P, Olivera MJ, Hernandez S, Lopez-Melgar B, Rojas-Gonzalez A, et al. Electrocardiogram and CMR to differentiate tachycardia-induced cardiomyopathy from dilated cardiomyopathy in patients admitted for heart failure. Heart Vessels. 2022;37(11):1850-1858.
doi pubmed - Xu Y, Li W, Wan K, Liang Y, Jiang X, Wang J, Mui D, et al. Myocardial tissue reverse remodeling after guideline-directed medical therapy in idiopathic dilated cardiomyopathy. Circ Heart Fail. 2021;14(1):e007944.
doi pubmed - Xu Y, Lin J, Liang Y, Wan K, Li W, Wang J, Zhu Y, et al. Prognostic value of left ventricular remodelling index in idiopathic dilated cardiomyopathy. Eur Heart J Cardiovasc Imaging. 2021;22(10):1197-1207.
doi pubmed - Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK. The diagnosis and evaluation of dilated cardiomyopathy. J Am Coll Cardiol. 2016;67(25):2996-3010.
doi pubmed - Authors/Task Force M, McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Bohm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2022;24(1):4-131.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.