

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Original Article
Volume 15, Number 4, August 2024, pages 233-245

Prevalence of Genetically Associated Dilated Cardiomyopathy: A Systematic Literature Review and Meta-Analysis
Michael C. Myersa, Su Wangb, Yue Zhonga, Sonomi Maruyamaa, Cindy Buenoa, Arnaud Bastiena, Mir Sohail Fazelib, Negar Golchina, c
aBristol Myers Squibb, Princeton, NJ, USA
bEvidinno Outcomes Research Inc., Vancouver, BC, Canada
cCorresponding Author: Negar Golchin, Bristol Myers Squibb, Princeton, NJ, USA
Manuscript submitted June 14, 2024, accepted July 30, 2024, published online August 15, 2024
Short title: Prevalence of Genetic DCM
doi: https://doi.org/10.14740/cr1680
| Abstract | ▴Top |
Background: Dilated cardiomyopathy (DCM) is a leading cause of heart failure and cardiac transplantation globally. Disease-associated genetic variants play a significant role in the development of DCM. Accurately determining the prevalence of genetically associated DCM (genetic DCM) is important for developing targeted prevention strategies. This review synthesized published literature on the global prevalence of genetic DCM across various populations, focusing on two of the most common variants: titin (TTN) and myosin heavy chain 7 (MYH7).
Methods: MEDLINE® and Embase were searched from database inception to September 19, 2022 for English-language studies reporting the prevalence of genetic DCM within any population. Studies using family history as a proxy for genetic DCM were excluded.
Results: Of 2,736 abstracts, 57 studies were included. Among the global adult or mixed (mostly adults with few pediatric patients) DCM population, median prevalence was 20.2% (interquartile range (IQR): 16.3-36.0%) for overall genetic DCM, 11.4% (IQR: 8.2-17.8%) for TTN-associated DCM, and 3.2% (IQR: 1.8-5.2%) for MYH7-associated DCM. Global prevalence of overall pediatric genetic DCM within the DCM population was similar (weighted mean: 21.3%). Few studies reported data on the prevalence of genetic DCM within the general population.
Conclusions: Our study identified variable prevalence estimates of genetic DCM across different populations and geographic locations. The current evidence may underestimate the genetic contributions due to limited screening and detection of potential DCM patients. Epidemiological studies using long-read whole genome sequencing to identify structural variants or non-coding variants are needed, as well as large cohort datasets with genotype-phenotype correlation analyses.
Keywords: Dilated cardiomyopathy; Genetics; Prevalence; Epidemiology
| Introduction | ▴Top |
Dilated cardiomyopathy (DCM) is a heterogeneous cardiac disorder where the predominant phenotypic description is left ventricular dilatation and systolic dysfunction, typically with left ventricular ejection fraction less than 45-50% [1-4]. Globally, it is one of the leading indicators of heart failure and heart transplantation in both children and adults [5].
DCM can have either a genetic or an acquired etiology, though they are not mutually exclusive. Familial and sporadic (no known family history of DCM) cases can occur, with or without genetic involvement. Disease-associated genetic variants are found in 20-35% of the familial DCM population [6]. The exact prevalence of disease-associated genetic variants in the sporadic DCM population is unclear [6, 7]. Currently, genetic variants in approximately 30 to 50 genes encoding sarcomeric, cytoskeletal, desmosomal, nuclear membrane, mitochondrial, and RNA-binding proteins have all been associated with DCM, possibly through polygenic combinatorial models [8-11]. Two of the most common sarcomeric genes frequently and definitively associated with DCM etiology are: titin (TTN) and myosin heavy chain 7 (MYH7) [10]. Protein truncating variants in TTN (TTNtv) account for around 25% of familial DCM and 18% of sporadic DCM [12]. Variants in MYH7 account for around 4% of familial DCM [8].
Prevalence estimates of DCM range from 1:250 up to 1:2,500 people within the general population [6, 8, 13]. The original estimate of 1:2,500 was based on a study conducted in Olmstead County Minnesota from 1975 to 1984 [14]. However, these results may be an underestimation due to under-reporting of the disease and less sophisticated diagnostic technology; most current estimates range between 1:250 and 500 [6, 8, 13]. In the United States in 2022, this roughly corresponds to 0.6 - 1.3 million people. Current prevalence estimates of DCM with associated genetic variants (referred to as genetic DCM, hereafter) may also be an underestimation due to limitations in disease diagnosis and genetic screening. Current definitions and criteria for diagnoses of DCM are unclear, leading to reduced genetic screening for DCM [2]. Furthermore, DCM-associated genetic variants have been observed within the general “healthy” population of those who have not been diagnosed with DCM [8, 10]. This demonstrates the incomplete penetrance of some DCM-associated genetic variants. The associated phenotypes could be subclinical or “asymptomatic” carriers resulting in under-reporting of clinical DCM patients. As well, most studies have focused on exonic variants. Far fewer studies have investigated the effects of intronic variants, mitochondrial deoxyribonucleic acid (DNA) variants, and structural variants on DCM etiology [11, 15]. Together, these factors complicate the ability to calculate the true prevalence of genetic DCM.
Despite numerous reviews on the genetic etiology of DCM, there has been no systematic literature review conducted on the prevalence of genetic DCM in the DCM population. We conducted a systematic literature review to assess the current landscape of evidence on the prevalence of adult and pediatric genetic-associated DCM (overall DCM-associated genetic variants, DCM-associated TTN variants, and DCM-associated MYH7 variants) in: 1) the clinical DCM population; 2) the general population; and 3) the subclinical DCM population. The prevalence of overall DCM-associated genetic variants in the healthy population was also presented.
| Materials and Methods | ▴Top |
This systematic review was performed according to standard methodologies for conducting and reporting systematic reviews as recommended by the Cochrane Handbook for Systematic Reviews of Interventions [16] and the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines [17].
Data sources and search strategies
Searches were carried out in MEDLINE® and Embase via OvidSP to capture records published from database inception to September 19, 2022 (Supplementary Materials 1 and 2, www.currentsurgery.org). Searches of relevant conferences and hand searches of reference lists of previously published literature reviews were also completed.
Study selection
Study eligibility criteria for inclusion in the systematic review were defined using the PICOS framework (Population, Intervention, Comparator, Outcome, Study design). Included were English-language studies reporting the prevalence of genetic DCM within any population. Studies using family history as a proxy for genetic DCM were excluded.
All abstracts were reviewed according to the PICOS criteria. Abstracts considered eligible for inclusion proceeded to a full-text screening phase, where they were screened by senior reviewers. Records deemed eligible following full-text screening were included for evidence synthesis.
Data extraction and quality assessment
Extracted data included study characteristics, patient characteristics (age of disease onset, sex, race/ethnicity, and comorbidities among genetic DCM patients), prevalence of genetic DCM, and genetic DCM sequencing methods. Quality of the included studies was assessed using the Newcastle-Ottawa scale for cohort studies [18] or the Joanna Briggs Institute checklist for cross-sectional studies [19].
Data analysis
Descriptive statistics were used to summarize data including measures of central tendency (mean and median) and variability (standard deviation (SD), interquartile range (IQR), and 95% confidence interval). For calculations with more than nine studies, median and IQR were chosen to account for outliers due to inter-study sequencing techniques and gene panel number variability. Medians were calculated by taking the median of all the studies’ values. For calculations with between three and nine studies (inclusive), weighted mean and weighted SD were chosen to account for the wide range of DCM sample size and outliers within a small sample size. Calculations were not carried out for values reported by less than three studies; instead, the full list of values were reported.
Prevalence of genetic DCM in the total DCM population was calculated by dividing the number of participants with clinically diagnosed DCM and DCM-associated genetic variants (overall, TTN, or MYH7) by the total number of participants with clinically diagnosed DCM (Equation 1). Prevalence of genetic DCM in the general population, and prevalence of DCM-associated genetic variants in healthy populations were calculated according to Equations 2 and 3, respectively. Overall genetic DCM prevalence was calculated by including studies that sequenced more than four genes. TTN-associated and MYH7-associated DCM prevalence estimates were calculated by including studies that sequenced TTN and MYH7, respectively. Subgroup prevalence analyses were conducted by geography, age, and genetic variant.
Forest plots were created to present the individual prevalence estimates per study as well as the overall median or weighted mean across all studies reporting. Confidence intervals on the proportions in the forest plots were calculated using the prop.test function for R, which uses Wilson’s score method [20].
| Results | ▴Top |
Study selection
In total, 2,720 abstracts were identified from Embase and MEDLINE®. Sixteen studies were added through hand-searching of published review papers [12, 21-37]. Following removal of duplicate records, most records were excluded during the initial abstract screening phase for reasons of study design (i.e., not an epidemiological study) or population (i.e., did not report on DCM). After full-text screening, 57 studies were included for qualitative synthesis (Fig. 1). Lists of the included studies and those excluded during full-text screening are provided in Supplementary Materials 3 and 4 (www.currentsurgery.org), respectively.
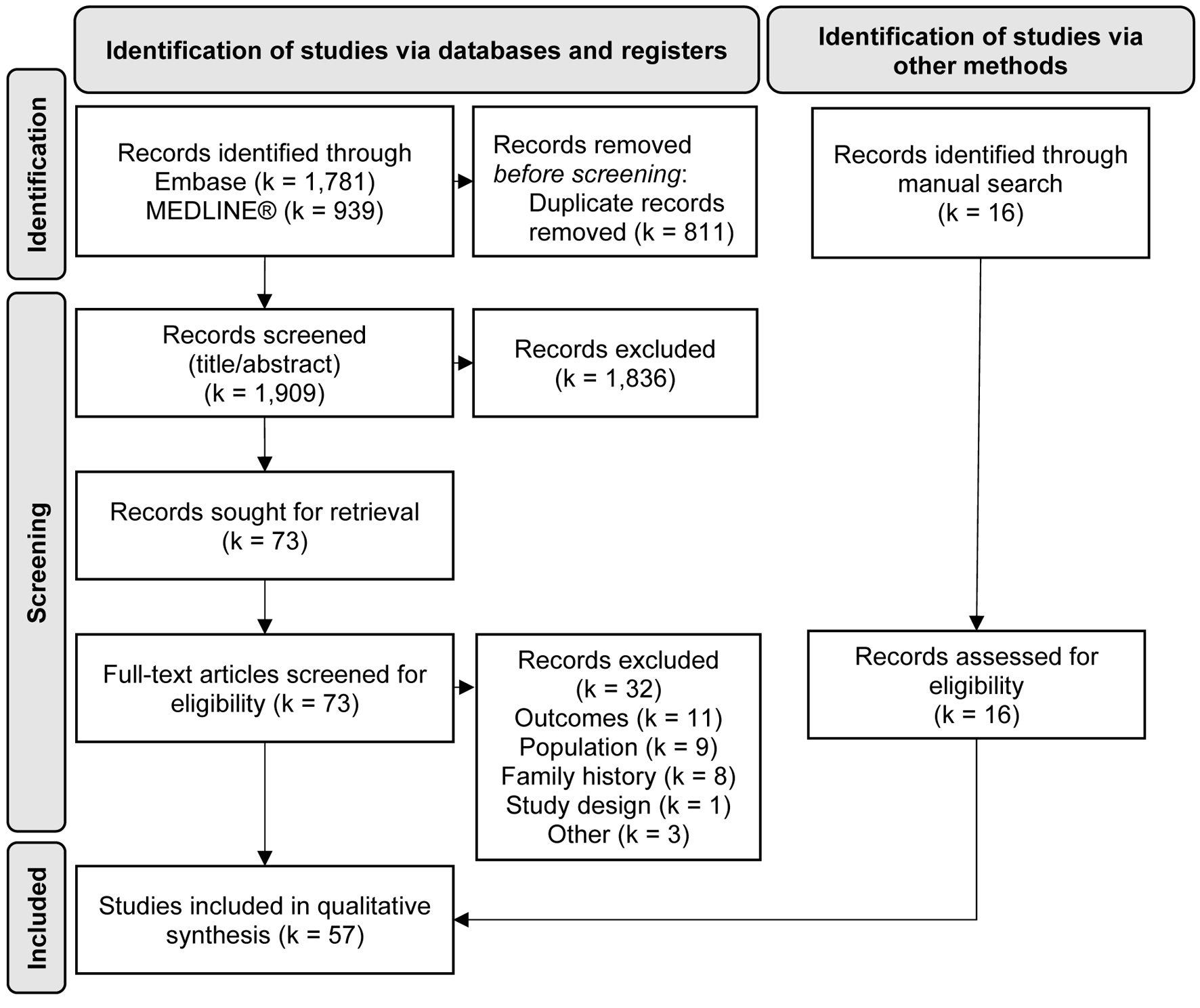 Click for large image | Figure 1. PRISMA flow diagram. k: number of records. |
Study characteristics, patient characteristics, and study quality
Of the 57 unique studies included, 31 were cross-sectional, 16 were prospective cohorts, and 10 were retrospective cohorts. The top three countries in which studies were conducted in were the United States (number of studies (k) = 8), the United Kingdom (UK; k = 6), and the Netherlands (k = 6). There were also eight multinational studies conducted in European countries, the United States, Australia, and Singapore (Supplementary Material 3, www.currentsurgery.org). The total sample size of the 57 included studies was 783,655 participants (median: 229; range: 21 [38] to 502,462 [39]). Three studies included participants from general populations with sample sizes of 71,313, 166,690, and 502,462 participants, respectively [39-41]. Forty-seven studies reported on adult populations, 10 reported on pediatric populations, and six reported on mixed adult and pediatric populations. Among studies reporting mixed populations, the large majority were adults. The median age of DCM onset was 45.5 years (range: 0.5 [42] to 57.1 [38] years). The median proportion of female participants was 36.6% (range: 10.7% [43] to 93.5% [44]). Seventeen studies reported race or ethnicity data; the majority of participants were White (median: 72.0%; range: 0% [26, 36, 38, 42, 45-48] to 100% [49, 50]). The top three most commonly reported comorbidities among participants were diabetes (k = 9; median: 13.9%) [12, 26, 41, 48, 51-55], hypertension (k = 8; median: 40.7%) [12, 41, 45, 48, 51-53, 55], and atrial fibrillation or flutter (k = 6; median: 26.6%) [22, 45, 47, 51, 53, 54].
The 26 cohort studies were generally of fair quality, with 24 studies receiving a final score between 4 and 6 (maximum of 9 points) on the Newcastle-Ottawa scale. The 31 cross-sectional studies were typically moderate to high quality. Twenty-one studies were considered low risk of bias in at least five of the eight domains of the Johanna Briggs Institute tool. Studies scoring lower were typically available as conference abstracts only at the time of this review (k = 5) with limited information on study methodology [40, 56-59]. However, it should be noted that upon comparing the estimates obtained from these abstracts with the prevalence estimates obtained from studies published as full journal articles at the time of this review, no large differences or outliers were detected. Therefore, inclusion of these conference abstracts in our evidence synthesis was considered justified.
Prevalence of genetic DCM
Forty-six studies reported on the prevalence of overall genetic DCM, 35 on the prevalence of TTN-associated DCM, and 28 on the prevalence of MYH7-associated DCM. Gene panels analyzed for overall genetic DCM were heterogeneous. Prevalence of genetic DCM was grouped by geography (global (including United States), or United States only), age (adults or mixed (adult and pediatric) population or pediatric only), and genetic variants (overall genetic DCM, TTN-associated DCM, or MYH7-associated DCM).
Overall genetic DCM
The prevalence of overall genetic DCM was similar between adults and children with DCM. Across the 36 studies that reported on adults/mixed populations with DCM globally, the median prevalence of overall genetic DCM was 20.2% (IQR: 16.3-36.0%; Table 1 [12, 21-41, 43-45, 47, 49-58, 60-70]; Fig. 2a). Similar global prevalence estimates were reported in pediatric populations with DCM (weighted mean: 21.3%, SD: 10.6%, k = 9; Table 2 [33, 34, 42, 54, 67, 71-75]; Fig. 3a). The prevalence estimates in the United States were similar for both the adult/mixed (weighted mean: 15.2%, SD: 2.5%, k = 3) and pediatric populations (weighted mean: 17.3%, SD: 4.9%, k = 3).
 Click to view | Table 1. Prevalence of Genetic DCM Within the Adult or Mixed Populations (Adults and Children) |
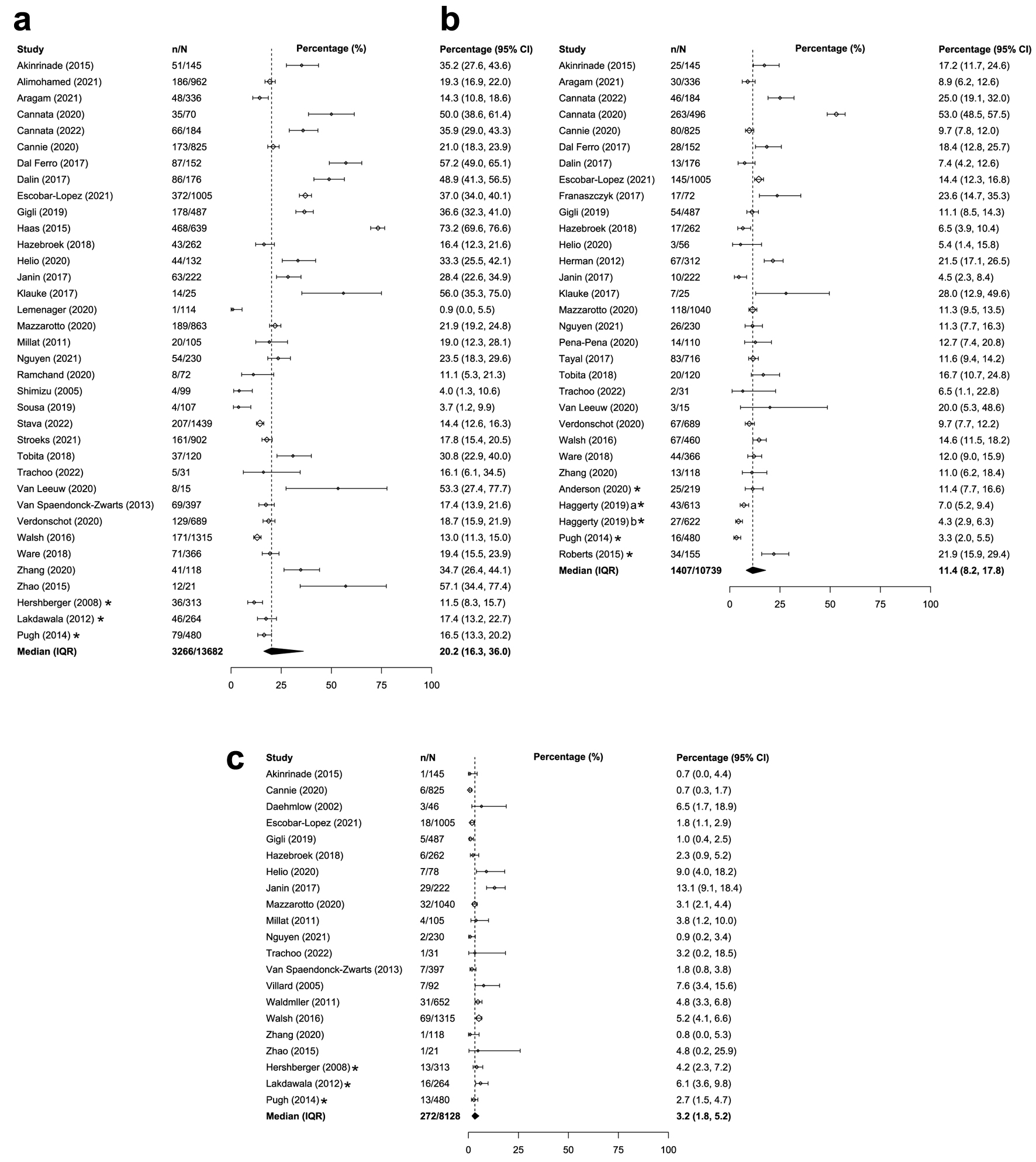 Click for large image | Figure 2. Median or weighted mean prevalence of overall genetic DCM in adults or adult/mixed populations (a), TTN-associated genetic DCM in adults or adult/mixed populations (b), and MYH7-associated genetic DCM in adults or adult/mixed populations (c). *Study was conducted only in the United States. aCalculated from the sample of DCM patients within the Penn Medicine BioBank. bCalculated from the sample of DCM patients within the Geisinger cohort. CI: confidence interval, calculated by reviewers; DCM: dilated cardiomyopathy; IQR: Interquartile range; n: numerator of calculation (e.g., number of overall genetic DCM patients); N: denominator of calculation (i.e., number of DCM patients); SD: weighted standard deviation. |
Click to view | Table 2. Prevalence of Genetic DCM Within the Pediatric DCM Population by Region |
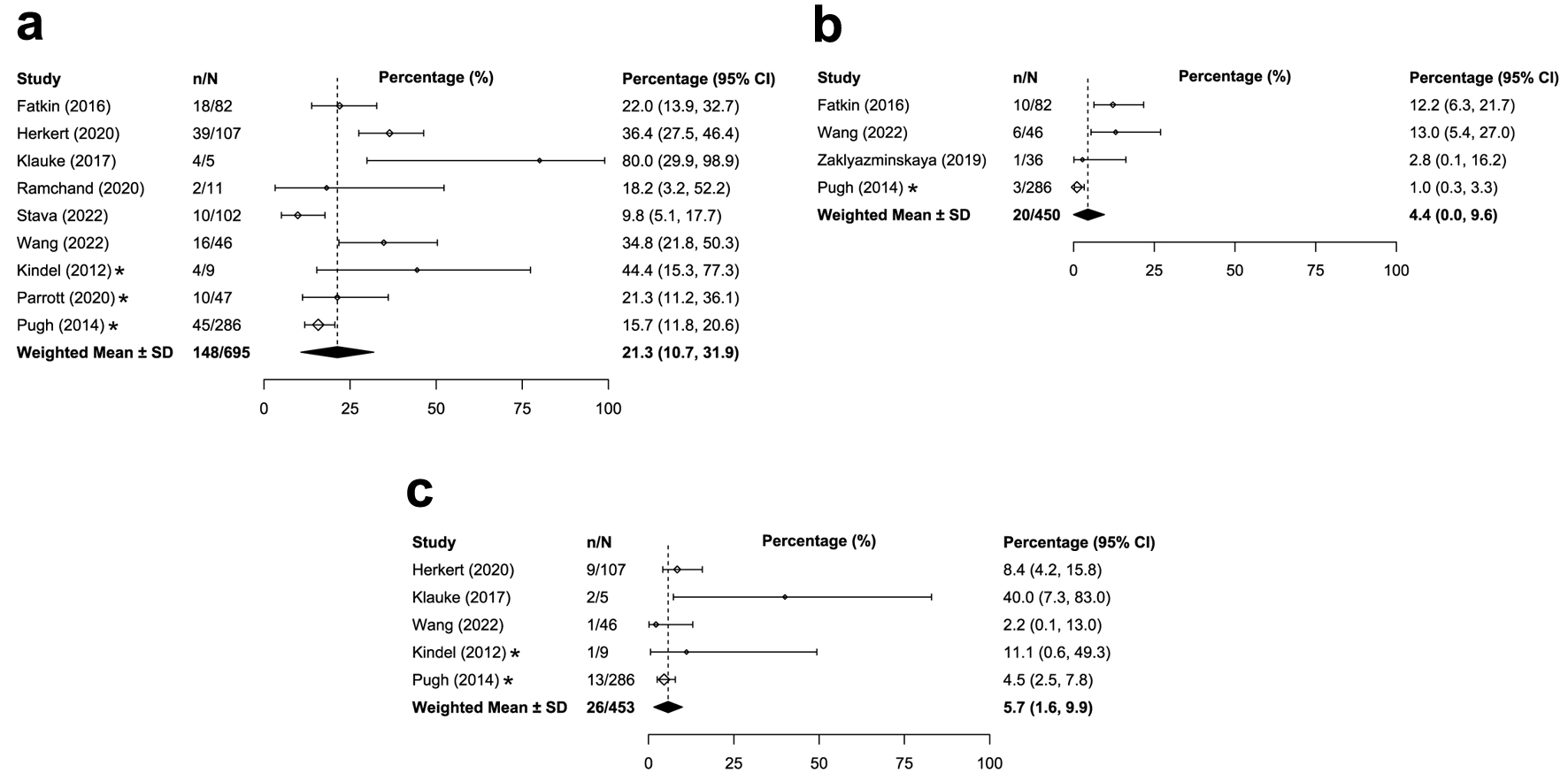 Click for large image | Figure 3. Median or weighted mean prevalence of overall genetic DCM in children (a), TTN-associated genetic DCM in children (b), and MYH7-associated genetic DCM in children (c). *Study was conducted only in the United States. CI: confidence interval, calculated by reviewers; DCM: dilated cardiomyopathy; IQR: interquartile range; n: numerator of calculation (e.g., number of overall genetic DCM patients); N: denominator of calculation (i.e., number of DCM patients); SD: weighted standard deviation. |
Two studies reported the prevalence of adults with genetic DCM within the general population using UK Biobank data. This biobank aims to represent the general UK population and includes clinical and genetic information of 500,000 volunteers from the UK recruited between 2006 and 2010 [76]. Aragam et al (2021) examined the data from 166,690 people in the UK Biobank and found 336 people with clinical DCM [40]. Of those 336 people with clinical DCM, 48 people had a pathogenic genetic variant in a DCM-associated gene. Using the UK Biobank data at different timepoints, Aragam et al (in 2021) reported that 0.029% of people in the UK general public have genetic DCM compared with 0.027% as reported by Shah et al (2022) [39].
TTN-associated DCM
The global prevalence of TTN-associated DCM was higher for adults compared with children with DCM. Among 30 studies reporting on adults/mixed populations with DCM, median prevalence of TTN-associated DCM was 11.4% (IQR: 8.2-17.8%; Table 1; Fig. 2b). Among children with DCM, weighted mean prevalence of TTN-associated DCM was 4.4% (SD: 5.1%, k = 4; Table 2; Fig. 3b). Prevalence estimates of TTN-associated DCM within the United States populations were similar for both adults (weighted mean: 6.9%; SD: 4.9%; k = 4) and children (1.0%; k = 1).
Only one study reported the prevalence of TTN-associated DCM among the general population. Haggerty et al (2019) [41] reported the prevalence of DCM patients with pathogenic DCM-associated TTN variants within two general populations: Geisinger 0.04% (27 participants with TTN; 61,040 participants within Geisinger cohort) and Penn Medicine BioBank (PMBB) 0.42% (43 participants with TTN; 10,289 participants within PMBB cohort; Table 1).
MYH7-associated DCM
The prevalence estimates of MYH7-associated DCM were similar for adults compared with children with DCM. Across 21 studies that reported on adults/mixed populations with DCM, median global prevalence of MYH7-associated DCM was 3.2% (IQR: 1.8-5.2%; Table 1; Fig. 2c). Findings were similar for the pediatric population; the weighted mean prevalence of MYH7-associated DCM was 5.7% globally (SD: 4.2%, k = 5; Table 2; Fig. 3c). Prevalence of MYH7-associated DCM in the United States was generally consistent with the global estimates for both adults (weighted mean: 4.0%; SD: 1.4%; k = 3) and children (up to 11.1%; k = 2).
DCM-associated genetic variants in healthy individuals
The prevalence of pathogenic DCM-associated genetic variants within healthy individuals was reported by six studies [12, 21, 24, 25, 50, 60]. These data represent the number of healthy individuals who were identified to have DCM-associated genetic variants but did not present with DCM anatomically or symptomatically (i.e., healthy individuals). For overall DCM-associated genetic variants, two studies focused on healthy individuals to assess the prevalence of genetic variants in DCM-associated genes [50, 60]. These unrelated healthy individuals were either healthy reference individuals in the European ancestry cohort of the 1000 Genomes project or healthy volunteers participating in the UK Digital Heart Project [50, 60]. Dalin et al (2017) [60] reported that 38.4% of healthy people have DCM-associated predicted pathogenic variants (healthy individuals with variant n = 193; healthy individuals n = 503) compared with 2.7% as reported by Ware et al (2018) [50] (healthy individuals with variant n = 12; healthy individuals n = 445; Table 3 [12, 21, 24, 25, 50, 60]).
Click to view | Table 3. Prevalence of DCM-Associated Genetic Variants in Healthy Individuals by Region |
Six studies reported the global prevalence of pathogenic DCM-associated TTN variants within healthy individuals [12, 21, 24, 25, 50, 60]. The unrelated healthy individuals were either from healthy control populations or from healthy reference populations. The weighted mean prevalence was 0.9% (SD: 0.2%; Table 3).
Mazzarotto et al (2020) [25] and Walsh et al (2016) [24] reported that 1.4% (healthy individuals with pathogenic MYH7 variant n = 13; healthy controls n = 912) and 1.4% (general population with pathogenic MYH7 variant n = 845; general population n = 60,471) of healthy people have DCM-associated predicted pathogenic MYH7 variants, respectively (Table 3). Both the healthy controls and people in the general population were unrelated to the DCM population within each study. The prevalence of MYH7 variants was similar to that of the TTN variants in healthy individuals.
Genetic DCM in subclinical DCM
Two studies reported the prevalence of subclinical genetic DCM. Vissing et al (2021) reported that 35.07% of DCM patients have pre-symptomatic familial DCM [59]. Shah et al (2022) reported that 0.13% of subclinical DCM patients (defined as the fulfillment of cardiac magnetic resonance imaging criteria for DCM in the absence of a clinical history of DCM) have putative pathogenic mutations in DCM-associated genes [39].
| Discussion | ▴Top |
To our knowledge, this is the first systematic review to investigate the prevalence of genetic DCM within adult and pediatric populations. In the adult global DCM population, we found a median prevalence of 20.2% (IQR: 16.3-36.0%) for overall DCM-associated genetic variants, 11.4% (8.2-17.8%) for DCM-associated TTN variants, and 3.2% (1.8-5.2%) for DCM-associated MYH7 variants. The prevalences of TTN and MYH7 variants are similar to those presented in previous narrative reviews [10, 77]. Overall genetic DCM prevalence in the DCM population reported in the current study cannot be compared to prior reviews. Prior reviews separated reported prevalence estimates of genetic DCM in familial or sporadic DCM populations, rather than the total DCM population. Nevertheless, prevalence of genetic DCM has been estimated to account for up to 35% of cases [6]. Frequency of genetically associated sporadic DCM is not well defined [10]. Overall DCM-associated genetic variant prevalence in our report could be underestimated as seven studies [38, 44, 46, 47, 61-63] excluded TTN from their cardiomyopathy gene panels, and five studies [56-59, 64] did not explicitly state the sequenced genes set. Excluding TTN from the gene panels could lead to overall genetic association underestimation since TTN variants comprise a large proportion of overall DCM-associated genetic variants [10, 12].
Similar prevalence results were found in the adult DCM population within the United States for overall DCM-associated genetic variants and TTN variants. The prevalence of the MYH7 variant in the adult DCM population (4.0%) within the United States was also similar to the global median (3.2%). Though, comparison should be carefully interpreted since only three studies reported prevalence within the United States [31, 34, 44]. All genetic variants (overall, TTN, MYH7) prevalence in the DCM population were similar between adults and children with DCM.
We also identified prevalence of genetic DCM in the adult general population. Globally, 0.029% [40] and 0.027% [39] of the UK Biobank population had genetic DCM (overall genetic variants). Data were analyzed at different timepoints between the two studies. The advantage of the UK Biobank is that it includes clinical and genetic information from half a million volunteers from the UK, aged 40 to 69, recruited between 2006 and 2010 [76]. However, “healthy volunteer” selection bias has been reported in the UK Biobank: Biobank participants were less likely to be obese, to smoke, and to drink alcohol on a daily basis and had fewer self-reported health conditions, compared with the general population [78]. The discrepancy in prevalence estimates observed across the two studies reporting on UK Biobank data at different time points could be due to differences in pathogenic variant determination. Aragam et al (2021) focused on monogenic, predicted loss of function variants in 91 clinical testing panel DCM genes [40], whereas Shah et al (2022) studied predicted pathogenic variants in 44 ClinGen-curated DCM genes [39]. However, the study by Aragam et al (2021) was only available as a conference abstract at the time of this review; thus, results may require re-evaluation upon the availability of peer-reviewed publications with further details into the methodology of calculating these estimates.
Only one study by Haggerty et al (2019) conducted in the United States reported the prevalence of genetic DCM for TTN variants in two general populations: 0.04% (Geisinger population) and 0.42% (PMBB population) [41]. These prevalence values may be an overestimation, especially compared with the prevalence of overall genetic variants in the general population. This could be due to differences in participant demographics. The UK Biobank aimed to represent the general population, compared with the Geisinger or PMBB populations, which represented general clinical and tertiary care populations in the Pennsylvania region. Geisinger participants were recruited from a general clinical population while PMBB participants were from a tertiary care facility where a larger proportion of the participants have more advanced diseases. Furthermore, a higher proportion of PMBB participants were of African ancestry compared to Geisinger, 21% versus 2.8%, perhaps suggesting ethnic differences in TTN variant burden.
Our review also identified evidence of a small proportion (2.7% [50]) of people with DCM-associated genetic variants who are healthy and do not present with clinical DCM symptoms. This indicates an incomplete penetrance mechanism of variant pathogenicity. DCM-associated variants can be tolerated in some healthy individuals without developing clinical DCM. However, an individual is over seven times more likely to develop DCM if they have a DCM-associated variant. This result is similarly found in DCM-associated TTN (> 10 times more likely) and MYH7 (> 2 times more likely) variants. Interestingly, Dalin et al (2017) reported that 38.4% of healthy people have DCM-associated genetic variants [60]. The high prevalence of genetic variants in DCM-associated genes in healthy individuals is mostly due to the inclusion of missense mutations (single protein changing DNA nucleotide mutation) in the study’s pathogenic variant inclusion criteria. This type of non-synonymous mutation is not as detrimental to the protein function compared with other non-synonymous mutations since only one protein amino acid is changed. Most studies do not include missense mutations in determining pathogenic variants. Excluding missense mutations from Dalin et al (2017), other non-synonymous mutations account for 1.2% of the healthy population, similar to the results reported in Ware et al (2018) [50]. No studies using data from children with DCM reported prevalence of genetic DCM in the general population or DCM-associated genetic variants in healthy populations.
Very few studies reported the prevalence of subclinical genetic DCM. In a study reporting the prevalence of overall genetic variants in pre-symptomatic familial DCM (35.07% [59]), the prevalence was higher than that reported in symptomatic DCM patients. This supports the hypothesis that DCM is a complex disease with heterogeneous genetic and environmental factors [8]. These results combined with the prevalence of DCM-associated genetic variants within the healthy population support the hypothesis that some of the predicted genetic variants may be required to occur in combination with other genetic variants and environmental stressors to determine disease pathogenesis, otherwise leading to unaffected carriers or subclinical disease presentations.
Lastly, previous reviews have discussed the underlying genetic mechanisms of DCM and how these mechanisms link to therapies [10]. As our understanding of these mechanisms evolves, subtypes of DCM can be further refined, and clinical management can ultimately be improved. Specifically, there is much opportunity in DCM for personalized medicine including therapies targeted for specific pathogenetic variants. Such targeted approaches can lead to more effective treatments with potentially fewer side effects. Taken together with our findings that DCM often has a substantial genetic component, and a significant portion of genetic carriers are often subclinical, further research should aim to investigate new therapies specific to different mechanisms and DCM-associated genes, as well as determine the most effective and safe treatments for pre-clinical asymptomatic DCM gene carriers.
Our review had several strengths. The literature searches in our review were comprehensive and included two major electronic databases, as well as several gray literature sources and hand-searching. Additionally, our evidence base included 57 studies from over 22 countries, thus should be representative of the current published evidence. There are also several limitations to our review. Our review is limited to only studies published in English, which may introduce selection bias against studies from non-English-speaking countries. This bias is likely minimal as many studies in this review enrolled patients from countries outside of the United States and Europe including China (n = 5), Japan (n = 2), Singapore (n = 1), Thailand (n = 1), and Vietnam (n = 1; Supplementary Material 3, www.currentsurgery.org). Findings on subclinical patients should be cautiously interpreted due to the low number of studies. As well, all studies that used genetic sequencing only focused on the protein coding exon region which accounts for only 2.8% of the human genome [79]. However, non-protein coding variants have been implicated in several human diseases [80]. By only sequencing the coding region, the current literature may underestimate the prevalence of genetic determinants in DCM etiology. Finally, the majority of studies only reported genetic variants that are predicted to be pathogenic according to the 2015 American College of Medical Genetics and Genomics guidelines and standards [81]. This disregards the “variants of unknown significance” which accounts for about 28.6% of individuals with cardiomyopathies, again possibly leading to underestimation of the prevalence of the genetic component in DCM [82]. This limitation in the current literature points to the need for more accurate ways to identify the subset of variants of unknown significance that are truly deleterious through large scale genotype-phenotype correlation studies and complementary functional studies.
Conclusions
Globally, a significant proportion of adult patients with DCM have genetic factors contributing to disease etiology; more than half is comprised of variants in the TTN gene and around one-sixth is comprised of variants in the MYH7 gene. However, the current literature may present an underestimation of overall genetic factors due to underdiagnosis of earlier stages of DCM, limited widespread genetic screening in DCM patients, and lack of characterization of non-protein coding genetic variant functions. Epidemiological studies using long-read whole genome sequencing and large cohort datasets with genotype-phenotype correlation analyses and complementary functional studies are needed.
| Supplementary Material | ▴Top |
Suppl 1. Search strategy for Embase (via OvidSP).
Suppl 2. Search strategy for MEDLINE® (via OvidSP).
Suppl 3. List of studies included in the review.
Suppl 4. List of excluded studies after full-text screening.
Acknowledgments
The authors would like to thank Kimberly Hofer, Warisha Usmani, and Nishu Gaind for their assistance with the data collection and medical writing. The authors would like to thank Dr. Jean-Paul Collet for his intellectual input into the analysis of the data and writing of this manuscript.
Financial Disclosure
This study was funded by Bristol Myers Squibb.
Conflict of Interest
MCM, YZ, SM, CB, AB, and NG are employees and/or shareholders of Bristol Myers Squibb. SW and MSF are or were employed by Evidinno Outcomes Research Inc. (Vancouver, BC, Canada) at the time of this study, which was contracted by Bristol Myers Squibb to conduct this study.
Informed Consent
Not applicable.
Author Contributions
Michael C. Myers: conceptualization, writing - review and editing, visualization, and supervision. Su Wang: investigation, validation, writing - original draft, writing - review and editing, and visualization. Yue Zhong: writing - review and editing, visualization, and supervision. Sonomi Maruyama: writing - review and editing, visualization, and supervision. Cindy Bueno: writing - review and editing, visualization, and supervision. Arnaud Bastien: writing - review and editing, visualization, and supervision. Mir Sohail Fazeli: conceptualization, methodology, writing - review and editing, visualization, and supervision. Negar Golchin: conceptualization, writing - review and editing, visualization, and supervision.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
DCM: dilated cardiomyopathy; DNA: deoxyribonucleic acid; IQR: interquartile range; MYH7: myosin heavy chain 7; PICOS: Population, Intervention, Comparator, Outcome, Study design; PMBB: Penn Medicine BioBank; PRISMA: Preferred Reporting Items for Systematic Reviews and Meta-Analyses; SD: standard deviation; TTN: titin; TTNtv: titin truncating variant; UK: United Kingdom
| References | ▴Top |
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270-276.
doi pubmed - Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Bohm M, Duboc D, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37(23):1850-1858.
doi pubmed - Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45(7):969-981.
doi pubmed - Mestroni L, Maisch B, McKenna WJ, Schwartz K, Charron P, Rocco C, Tesson F, et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J. 1999;20(2):93-102.
doi pubmed - Taylor DO, Edwards LB, Boucek MM, Trulock EP, Aurora P, Christie J, Dobbels F, et al. Registry of the International Society for Heart and Lung Transplantation: twenty-fourth official adult heart transplant report—2007. J Heart Lung Transplant. 2007;26(8):769-781.
doi pubmed - Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. 2017;390(10092):400-414.
doi pubmed - Ganesh SK, Arnett DK, Assimes TL, Basson CT, Chakravarti A, Ellinor PT, Engler MB, et al. Genetics and genomics for the prevention and treatment of cardiovascular disease: update: a scientific statement from the American Heart Association. Circulation. 2013;128(25):2813-2851.
doi pubmed - Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531-547.
doi pubmed - McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123(1):19-26.
doi pubmed pmc - McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. 2017;121(7):731-748.
doi pubmed pmc - Tayal U, Ware JS, Lakdawala NK, Heymans S, Prasad SK. Understanding the genetics of adult-onset dilated cardiomyopathy: what a clinician needs to know. Eur Heart J. 2021;42(24):2384-2396.
doi pubmed pmc - Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619-628.
doi pubmed pmc - Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: A translational review of current literature. J Intern Med. 2019;286(4):362-372.
doi pubmed - Codd MB, Sugrue DD, Gersh BJ, Melton LJ, 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation. 1989;80(3):564-572.
doi pubmed - Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010;12(11):655-667.
doi pubmed pmc - Higgins J, Green S. Handbook for systematic reviews of interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration. 2011. http://handbook.cochrane.org.
- Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, Shamseer L, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.
doi pubmed pmc - Wells GA, Shea B, O’Connell D, Peterson J, Welch V, Losos M, Tugwell P. In: The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. Oxford; 2000.
- The Joanna Briggs Institute. The Joanna Briggs Institute Critical Appraisal tools for use in JBI Systematic Reviews: Checklist for Analytical Cross Sectional Studies. 2017; https://jbi.global/sites/default/files/2019-05/JBI_Critical_Appraisal-Checklist_for_Analytical_Cross_Sectional_Studies2017_0.pdf.
- Newcombe RG. Two-sided confidence intervals for the single proportion: comparison of seven methods. Stat Med. 1998;17(8):857-872.
doi pubmed - Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7(270):270ra276.
doi pubmed pmc - Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenpera P, Koillinen H, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36(34):2327-2337.
doi pubmed pmc - Escobar-Lopez L, Ochoa JP, Mirelis JG, Espinosa MA, Navarro M, Gallego-Delgado M, Barriales-Villa R, et al. Association of genetic variants with outcomes in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol. 2021;78(17):1682-1699.
doi pubmed - Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19(2):192-203.
doi pubmed pmc - Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, Govind R, et al. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation. 2020;141(5):387-398.
doi pubmed pmc - Zhang XL, Xie J, Lan RF, Kang LN, Wang L, Xu W, Xu B. Genetic basis and genotype-phenotype correlations in han chinese patients with idiopathic dilated cardiomyopathy. Sci Rep. 2020;10(1):2226.
doi pubmed pmc - Daehmlow S, Erdmann J, Knueppel T, Gille C, Froemmel C, Hummel M, Hetzer R, et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;298(1):116-120.
doi pubmed - Dal Ferro M, Stolfo D, Altinier A, Gigli M, Perrieri M, Ramani F, Barbati G, et al. Association between mutation status and left ventricular reverse remodelling in dilated cardiomyopathy. Heart. 2017;103(21):1704-1710.
doi pubmed - Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, Stolfo D, et al. Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2019;74(11):1480-1490.
doi pubmed pmc - Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36(18):1123-1135a.
doi pubmed - Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, et al. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1(1):21-26.
doi pubmed pmc - Janin A, N'Guyen K, Habib G, Dauphin C, Chanavat V, Bouvagnet P, Eschalier R, et al. Truncating mutations on myofibrillar myopathies causing genes as prevalent molecular explanations on patients with dilated cardiomyopathy. Clin Genet. 2017;92(6):616-623.
doi pubmed - Klauke B, Gaertner-Rommel A, Schulz U, Kassner A, Zu Knyphausen E, Laser T, Kececioglu D, et al. High proportion of genetic cases in patients with advanced cardiomyopathy including a novel homozygous Plakophilin 2-gene mutation. PLoS One. 2017;12(12):e0189489.
doi pubmed pmc - Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16(8):601-608.
doi pubmed - Tayal U, Newsome S, Buchan R, Whiffin N, Halliday B, Lota A, Roberts A, et al. Phenotype and clinical outcomes of titin cardiomyopathy. J Am Coll Cardiol. 2017;70(18):2264-2274.
doi pubmed pmc - Tobita T, Nomura S, Fujita T, Morita H, Asano Y, Onoue K, Ito M, et al. Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. Sci Rep. 2018;8(1):1998.
doi pubmed pmc - Villard E, Duboscq-Bidot L, Charron P, Benaiche A, Conraads V, Sylvius N, Komajda M. Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur Heart J. 2005;26(8):794-803.
doi pubmed - Zhao Y, Feng Y, Zhang YM, Ding XX, Song YZ, Zhang AM, Liu L, et al. Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int J Mol Med. 2015;36(6):1479-1486.
doi pubmed pmc - Shah RA, Asatryan B, Sharaf Dabbagh G, Aung N, Khanji MY, Lopes LR, van Duijvenboden S, et al. Frequency, penetrance, and variable expressivity of dilated cardiomyopathy-associated putative pathogenic gene variants in UK biobank participants. Circulation. 2022;146(2):110-124.
doi pubmed pmc - Aragam KG, Biddinger K, Jurgens SJ, Wang M, Pirruccello J, Maamari D, Chaffin M, et al. Combined assessments of monogenic and polygenic risk for dilated cardiomyopathy. Circulation Conference: American Hearts Association's. 2021;144(SUPPL 1):A13271.
- Haggerty CM, Damrauer SM, Levin MG, Birtwell D, Carey DJ, Golden AM, Hartzel DN, et al. Genomics-first evaluation of heart disease associated with titin-truncating variants. Circulation. 2019;140(1):42-54.
doi pubmed pmc - Wang Y, Han B, Fan Y, Yi Y, Lv J, Wang J, Yang X, et al. Next-generation sequencing reveals novel genetic variants for dilated cardiomyopathy in pediatric Chinese patients. Pediatr Cardiol. 2022;43(1):110-120.
doi pubmed - Waldmuller S, Erdmann J, Binner P, Gelbrich G, Pankuweit S, Geier C, Timmermann B, et al. Novel correlations between the genotype and the phenotype of hypertrophic and dilated cardiomyopathy: results from the German Competence Network Heart Failure. Eur J Heart Fail. 2011;13(11):1185-1192.
doi pubmed - Lakdawala NK, Funke BH, Baxter S, Cirino AL, Roberts AE, Judge DP, Johnson N, et al. Genetic testing for dilated cardiomyopathy in clinical practice. J Card Fail. 2012;18(4):296-303.
doi pubmed pmc - Nguyen TV, Tran Vu MT, Do TNP, Tran THN, Do TH, Nguyen TMH, Tran Huynh BN, et al. Genetic determinants and genotype-phenotype correlations in Vietnamese patients with dilated cardiomyopathy. Circ J. 2021;85(9):1469-1478.
doi pubmed - Shen C, Xu L, Sun X, Sun A, Ge J. Genetic variants in Chinese patients with sporadic dilated cardiomyopathy: a cross-sectional study. Ann Transl Med. 2022;10(3):129.
doi pubmed pmc - Shimizu M, Ino H, Yasuda T, Fujino N, Uchiyama K, Mabuchi T, Konno T, et al. Gene mutations in adult Japanese patients with dilated cardiomyopathy. Circ J. 2005;69(2):150-153.
doi pubmed - Xiao L, Wu D, Sun Y, Hu D, Dai J, Chen Y, Wang D. Whole-exome sequencing reveals genetic risks of early-onset sporadic dilated cardiomyopathy in the Chinese Han population. Sci China Life Sci. 2022;65(4):770-780.
doi pubmed - Franaszczyk M, Chmielewski P, Truszkowska G, Stawinski P, Michalak E, Rydzanicz M, Sobieszczanska-Malek M, et al. Titin truncating variants in dilated cardiomyopathy - prevalence and genotype-phenotype correlations. PLoS One. 2017;12(1):e0169007.
doi pubmed pmc - Ware JS, Amor-Salamanca A, Tayal U, Govind R, Serrano I, Salazar-Mendiguchia J, Garcia-Pinilla JM, et al. Genetic etiology for alcohol-induced cardiac toxicity. J Am Coll Cardiol. 2018;71(20):2293-2302.
doi pubmed pmc - Anderson JL, Christensen GB, Escobar H, Horne BD, Knight S, Jacobs V, Afshar K, et al. Discovery of titin gene truncating variant mutations and 5-year outcomes in patients with nonischemic dilated cardiomyopathy. Am J Cardiol. 2020;137:97-102.
doi pubmed - Hazebroek MR, Krapels I, Verdonschot J, van den Wijngaard A, Vanhoutte E, Hoos M, Snijders L, et al. Prevalence of pathogenic gene mutations and prognosis do not differ in isolated left ventricular dysfunction compared with dilated cardiomyopathy. Circ Heart Fail. 2018;11(3):e004682.
doi pubmed - Helio T, Elliott P, Koskenvuo JW, Gimeno JR, Tavazzi L, Tendera M, Kaski JP, et al. ESC EORP Cardiomyopathy Registry: real-life practice of genetic counselling and testing in adult cardiomyopathy patients. ESC Heart Fail. 2020;7(5):3013-3021.
doi pubmed pmc - Ramchand J, Wallis M, Macciocca I, Lynch E, Farouque O, Martyn M, Phelan D, et al. Prospective Evaluation of the Utility of Whole Exome Sequencing in Dilated Cardiomyopathy. J Am Heart Assoc. 2020;9(2):e013346.
doi pubmed pmc - Verdonschot JAJ, Hazebroek MR, Krapels IPC, Henkens M, Raafs A, Wang P, Merken JJ, et al. Implications of genetic testing in dilated cardiomyopathy. Circ Genom Precis Med. 2020;13(5):476-487.
doi pubmed - Cannata A, Merlo M, Manca P, Dal Ferro M, Paldino A, Artico J, Gentile P, et al. The late-onset dilated cardiomyopathy. Eur Heart J. 2020;41(SUPPL 2):2096.
- Cannie DE, Protonotarios A, Lorenzini M, Akhtar M, Syrris P, Lopes L, Elliott P. The influence of age on the diagnostic yield of genetic testing in dilated cardiomyopathy. Eur Heart J. 2020;41(SUPPL 2):2102.
- Van Leeuw JB, Gillardin AF, Vo C, Beauloye C, Gerber B, De Waroux JBLP, Marchandise S, et al. Prevalence and nature of genetic mutations identified in patients with cardiac diseases referred to a specialised consultation. Acta Clin Belg. 2020;75(Supplement 1):55.
- Vissing CR, Espersen K, Mills HL, Jurlander R, Vinther Skriver SV, Thune JJ, Raja AA, et al. Family screening in dilated cardiomyopathy; long-term incidence and potential for limiting follow-up. Circulation Conference: American Hearts Association's. 2021;144(SUPPL 1)A11176.
- Dalin MG, Engstrom PG, Ivarsson EG, Unneberg P, Light S, Schaufelberger M, Gilljam T, et al. Massive parallel sequencing questions the pathogenic role of missense variants in dilated cardiomyopathy. Int J Cardiol. 2017;228:742-748.
doi pubmed - Millat G, Bouvagnet P, Chevalier P, Sebbag L, Dulac A, Dauphin C, Jouk PS, et al. Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur J Med Genet. 2011;54(6):e570-575.
doi pubmed - Sousa A, Canedo P, Azevedo O, Lopes L, Pinho T, Baixia M, Rocha-Goncalves F, et al. Molecular characterization of Portuguese patients with dilated cardiomyopathy. Rev Port Cardiol (Engl Ed). 2019;38(2):129-139.
doi pubmed - van Spaendonck-Zwarts KY, van Rijsingen IA, van den Berg MP, Lekanne Deprez RH, Post JG, van Mil AM, Asselbergs FW, et al. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: overview of 10 years' experience. Eur J Heart Fail. 2013;15(6):628-636.
doi pubmed - Lemenager P, Franck YK, Corlin F, Bouscaren N, Nacher M, Adenis A. Aetiological and morphological spectrum of cardiomyopathies in French Guiana: a retrospective study. Open Heart. 2020;7(1):e001206.
doi pubmed pmc - Alimohamed MZ, Johansson LF, Posafalvi A, Boven LG, van Dijk KK, Walters L, Vos YJ, et al. Diagnostic yield of targeted next generation sequencing in 2002 Dutch cardiomyopathy patients. Int J Cardiol. 2021;332:99-104.
doi pubmed - Cannata A, Merlo M, Dal Ferro M, Barbati G, Manca P, Paldino A, Graw S, et al. Association of titin variations with late-onset dilated cardiomyopathy. JAMA Cardiol. 2022;7(4):371-377.
doi pubmed pmc - Stava TT, Leren TP, Bogsrud MP. Molecular genetics in 4408 cardiomyopathy probands and 3008 relatives in Norway: 17 years of genetic testing in a national laboratory. Eur J Prev Cardiol. 2022;29(13):1789-1799.
doi pubmed - Stroeks S, Hellebrekers D, Claes GRF, Tayal U, Krapels IPC, Vanhoutte EK, van den Wijngaard A, et al. Clinical impact of re-evaluating genes and variants implicated in dilated cardiomyopathy. Genet Med. 2021;23(11):2186-2193.
doi pubmed pmc - Trachoo O, Yingchoncharoen T, Ngernsritrakul T, Iemwimangsa N, Panthan B, Klumsathien S, Srisukh S, et al. Molecular genetic testing for hypertrophic and dilated cardiomyopathy in inherited cardiovascular condition genetics service: lessons from a Thai cohort. Eur J Med Genet. 2022;30(SUPPL 1):191.
- Pena-Pena ML, Ochoa JP, Barriales-Villa R, Cicerchia M, Palomino-Doza J, Salazar-Mendiguchia J, Lamounier A, et al. Prognostic implications of pathogenic truncating variants in the TTN gene. Int J Cardiol. 2020;316:180-183.
doi pubmed - Fatkin D, Lam L, Herman DS, Benson CC, Felkin LE, Barton PJR, Walsh R, et al. Titin truncating mutations: A rare cause of dilated cardiomyopathy in the young. Prog Pediatr Cardiol. 2016;40:41-45.
- Herkert JC, Van Der Meulen MH, Den Boer SL, Du Marchie Sarvaas GJ, Blom N, Ten Harkel ADJ, Breur HMPJ, et al. Utility of genetics for risk stratification in paediatric dilated cardiomyopathy. Eur J Med Genet. 2020;28(SUPPL 1):267-268.
- Kindel SJ, Miller EM, Gupta R, Cripe LH, Hinton RB, Spicer RL, Towbin JA, et al. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail. 2012;18(5):396-403.
doi pubmed pmc - Parrott A, Khoury PR, Shikany AR, Lorts A, Villa CR, Miller EM. Investigation of de novo variation in pediatric cardiomyopathy. Am J Med Genet C Semin Med Genet. 2020;184(1):116-123.
doi pubmed - Zaklyazminskaya E, Mikhailov V, Bukaeva A, Kotlukova N, Povolotskaya I, Kaimonov V, Dombrovskaya A, et al. Low mutation rate in the TTN gene in paediatric patients with dilated cardiomyopathy - a pilot study. Sci Rep. 2019;9(1):16409.
doi pubmed pmc - Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779.
doi pubmed pmc - Kayvanpour E, Sedaghat-Hamedani F, Amr A, Lai A, Haas J, Holzer DB, Frese KS, et al. Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol. 2017;106(2):127-139.
doi pubmed - Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, Collins R, et al. Comparison of sociodemographic and health-related characteristics of UK biobank participants with those of the general population. Am J Epidemiol. 2017;186(9):1026-1034.
doi pubmed pmc - Rigau M, Juan D, Valencia A, Rico D. Intronic CNVs and gene expression variation in human populations. PLoS Genet. 2019;15(1):e1007902.
doi pubmed pmc - Zhang F, Lupski JR. Non-coding genetic variants in human disease. Hum Mol Genet. 2015;24(R1):R102-110.
doi pubmed pmc - Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424.
doi pubmed pmc - Fatkin D, Johnson R. Variants of uncertain significance and "Missing Pathogenicity". J Am Heart Assoc. 2020;9(3):e015588.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.