

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Review
Volume 15, Number 5, October 2024, pages 330-339

The Role of Inducible Nitric Oxide Synthase in Assessing the Functional Level of Coronary Artery Lesions in Chronic Coronary Syndrome
Admina Senderovica, b, Semira Galijasevicb, c
aPublic Institution of Health Centers of the Canton of Sarajevo, Laboratory Diagnostics Service of the Ilidza, Health Center, Ilidza, Bosnia and Herzegovina
bSarajevo Medical School, University Sarajevo School of Science and Technology, Sarajevo, Bosnia and Hercegovina
cCorresponding Author: Semira Galijasevic, Sarajevo Medical School, University Sarajevo School of Science and Technology, Sarajevo, Bosnia and Hercegovina
Manuscript submitted July 10, 2024, accepted September 16, 2024, published online October 11, 2024
Short title: Inflammation and Coronary Artery Lesions
doi: https://doi.org/10.14740/cr1700
- Abstract
- Introduction
- CCS, MPO, and Oxidative Stress
- Atherosclerosis Pathogenesis
- The Role of iNOS and MPO in the Development of Atherosclerosis
- Interactions and Correlative Links of Functional Level of Lesions and iNOS
- Functional Phenotypes of Coronary Atherosclerosis
- Conclusions
- References
| Abstract | ▴Top |
Chronic coronary syndrome (CCS) is a long-term manifestation of coronary artery disease, marked by stable but recurring chest pain and myocardial ischemia due to the gradual buildup of atherosclerotic plaques in the coronary arteries. It is a metabolic disorder of coronary arteries characterized by oxidative stress, endothelial dysfunction, inflammation, and hyperlipidemia. The imbalance in oxidative-antioxidative status contributes to stable ischemic heart disease. Oxidative stress involves reactive oxygen and nitrogen species, leading to low-density lipoprotein (LDL) oxidation. Endothelial dysfunction, marked by reduced nitric oxide (NO) bioavailability, is an early onset of CCS, affecting vasodilation, cell proliferation, and inflammatory responses. Enzyme myeloperoxidase (MPO), traditionally considered protective, plays a dual role in initiating and progressing inflammatory diseases. MPO interacts with NO, modulating its catalytic activity. Elevated NO levels inhibit MPO through a reversible complex formation, preventing NO-induced inhibition by inducible nitric oxide synthase (iNOS). MPO also inactivates endothelial nitric oxide synthase (eNOS) and reacts with L-arginine, hindering NO synthesis. The interplay between MPO and NO significantly influences inflammation sites, impacting peroxidation rates and oxidation reactions. Peroxynitrite, a reactive species, contributes to nitration of tyrosine residues and lipid peroxidation. Mechanistic pathways suggest MPO enhances iNOS catalytic activity, influencing CCS development. iNOS, implicated in inflammation and atherosclerosis, is connected to NO regulation. This review analyzes the complex interplay of MPO, iNOS, and NO that affects plaque morphology, oxidative stress, and inflammation, contributing to atherosclerosis progression. Therefore, it is possible that the phenotypes of atherosclerotic plaques, focal and diffuse coronary artery disease, could be defined by the relationship between MPO and iNOS.
Keywords: Functional level of lesions; Inflammation; Inducible nitric oxide synthase; Nitric oxide; Myeloperoxidase; Endothelial dysfunction; Coronary physiology; Immunometabolism
| Introduction | ▴Top |
Chronic coronary syndrome (CCS) represents a progressive and long-term manifestation of coronary artery disease (CAD). It is mainly primarily characterized by stable but recurring episodes of chest pain (angina) and myocardial ischemia. Its pathophysiology is deeply rooted in the gradual accumulation of atherosclerotic plaques within the coronary arteries, leading to these vessels’ progressive narrowing and hardening [1].
The molecular mechanism of CCS is linked to the progression of atherosclerosis, initiated by endothelial dysfunction and oxidative stress.
One of the major components of the development and progression of CCS is endothelial dysfunction. This dysfunction is often a consequence of factors such as hypertension, hyperlipidemia, diabetes, and smoking, which contribute to oxidative stress and inflammation within the vascular system. Oxidative stress, in particular, results from an imbalance between the production of reactive oxygen species (ROS) and the body’s antioxidant defenses [2]. Considering that the pro-inflammatory state is directly associated with increased ROS, it is believed that elevated ROS may play a crucial role in the normal aging process and the pathogenesis of numerous chronic diseases, including cancer, atherosclerosis, cardiovascular diseases, diabetes, Parkinson’s, and Alzheimer’s disease.
Produced ROS can directly affect endothelial cells and degrade the extracellular matrix, exacerbating the inflammatory response and promoting the progression of atherosclerosis. Additionally, ROS species through the different mechanisms can modify lipoproteins, making them more atherogenic [3]. ROS generated by peroxidase enzymes not only damages endothelial cells but also reduces the bioavailability of nitric oxide (NO), a crucial molecule for vasodilation [4]. NO is synthesized by endothelial nitric oxide synthase (eNOS), and its primary function is to maintain vascular tone and inhibit platelet aggregation and leukocyte adhesion. Oxidative stress disrupts NO synthesis and function, further impairing endothelial function and promoting vasoconstriction, thrombosis, and inflammation [5].
Control of the inflammatory response develops through several molecular pathways involving the interaction of inducible nitric oxide synthase (iNOS) with myeloperoxidase (MPO), NO, and eNOS. In this review, we focus on mechanisms that underscore the importance of oxidative stress and inflammation in the pathophysiology of CCS. Understanding these underlying mechanisms is crucial for developing strategies to manage and treat CCS, improving patient outcomes, and possible development of therapies.
| CCS, MPO, and Oxidative Stress | ▴Top |
CCS with metabolic disorders is defined as a stable disease of the coronary arteries. Essentially, it is a progressive and continuous process with pathophysiological changes in coronary blood vessels and the clinically silent nature of the disease [1]. CAD is associated with an imbalance between the oxidative-antioxidative status of the organism and inflammatory processes. Inflammatory activation and dysfunction of the endothelium are key events in the development and pathophysiology of atherosclerosis and are associated with an elevated risk of cardiovascular events [6].
Oxidative stress is mediated by intracellular production and accumulation of ROS and reactive nitrogen species (RNS).
ROS are chemically reactive oxygen-based intermediaries. The process of ROS metabolism involves the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which facilitates the transfer of electrons from NADPH to molecular oxygen, generating superoxide anions (O-2) as the primary free radical. Antioxidant scavenging enzymes, mostly superoxide dismutase (SOD), play a crucial role in the dismutation of superoxide radical (O•-2) into non-radical species, specifically hydrogen peroxide (H2O2), along with the action of catalase (CAT) that dismutase O•-2 into the water molecules [7]. In addition, glutathione peroxidase 1 (GPx1), another antioxidant enzyme, reduces H2O2 and soluble lipid hydroperoxides using glutathione (GSH) as a source of reducing power [8]. These enzymes play a vital role in protecting cells from oxidative damage and maintaining the integrity of the vascular system, thereby limiting damage to the host. Conversely, O•-2 can potentially transform into other ROS that can adversely affect nucleic acids, proteins, and cell membranes [8, 9]. Neutrophils produce significant levels of ROS at various cellular locations. The NADPH-oxidase enzyme complex, predominantly localized in specific and gelatinase granules, translocates upon stimulation to generate ROS at the membrane-bound b-cytochrome, separating them from cytoplasmic components. These ROS can be released extracellularly in response to chemoattractants, while during phagocytosis, ROS production occurs within phagolysosomes [10].
ROS can react with RNS, causing reduced bioavailability of NO and forming peroxynitrite species (ONOO-) that are even more damaging to the nearby tissue [11].
NO is an essential intracellular and intercellular biologically active molecule that acts on various physiological and pathophysiological functions in the body, including vasodilation, cell proliferation, antimicrobial defense, and regulation of inflammatory responses [12]. It is a short-lived molecule, highly reactive, and easily diffuses from the point of production. There are three isoforms of nitric oxide synthase (NOS) found within physiological systems: endothelial (eNOS), neuronal (neuronal nitric oxide synthase (nNOS)), and inducible (iNOS), each localized in various organs. The constitutive enzymes, eNOS and nNOS, rely on calcium and calmodulin and are primarily found in vascular endothelial cells. In contrast, the inducible form, iNOS, which operates independently of calcium, is found in macrophages and neutrophils [13].
Endothelial NOS is the most prevalent form, acting as a significant NO source under physiological conditions and yielding both beneficial and adverse effects depending on the environment and interacting molecules. Inducible NOS, on the other hand, is activated in response to cytokines and inflammation sites, distinguishing it from endothelial NOS, which is closely associated with endothelial cell membranes [5]. Both enzymes produce NO from the amino acid L-arginine, using oxygen and NADPH in the presence of the cofactor tetrahydrobiopterin (H4B). Disrupted NO metabolism, especially reduced production and bioavailability of NO, is recognized in the literature as a risk factor for the development of cardiometabolic diseases and as a key modulator of vascular diseases [14]. In inflammatory conditions, a significant increase in NO as an inflammatory mediator, in combination with ROS, contributes to oxidative stress.
MPO is an iron-containing heme protein, a member of the peroxidase subfamily, abundantly expressed in immune cells such as neutrophils, leukocytes, lymphocytes, monocytes, and macrophages. MPO is released from these cells during the process of oxidative burst. The granule-localized MPO plays a significant role in this context by converting H2O2 to hypochlorous acid (HOCl), enhancing the clearance of invading pathogens. Furthermore, MPO can directly convert O•-2 into singlet oxygen (1O2). Additionally, the presence of ferric iron can facilitate the conversion of O•-2 and H2O2 into the highly reactive hydroxyl radical (OH•-) [15].
When MPO is released into circulation, it gets adsorbed on negatively charged surfaces such as endothelium or lipoproteins. The presence of chlorinated apolipoprotein B-100 extracted from human lesion material indicates the direct involvement of the MPO-H2O2-Cl- system with low-density lipoprotein (LDL) as one of the main targets of damage in oxidative modifications, underscoring its potential role in the development of atherosclerosis. Concerning DNA, MPO is involved in oxidation-dependent damage that leads to single- or double-strand breaks, resulting in the formation of 8-oxo-2-deoxyguanosine (8-oxo-dG), which is not specific to MPO but is a common signature of ROS damage to nucleic acids [16]. The release and activity of MPO are directly associated with increased levels of oxidative stress with oxygen-reactive species and the activation of protein (MAP) signaling pathways, among other auxiliary pathways [17]. Increased MPO activity is linked to numerous pathologies with compelling evidence of initiating and progressing inflammatory events [18]. For example, leukocyte and MPO levels in serum are elevated in patients with CAD and can be used as markers for cardiovascular events [11]. Oxidants derived from MPO are generated in various diseases, implying the enzyme is a catalyst for oxidative reactions in the vascular wall [19].
The direct interaction between MPO and NO reveals that NO modulates MPO’s catalytic activity through specific mechanisms. NO expedites both the formation and decay of compound II, the rate-limiting step in the peroxidase cycle. Higher NO levels lead to reversible MPO inhibition through the creation of the MPO-Fe(III)-NO complex, acting as both a ligand and substrate for MPO. Additionally, MPO prevents NO-induced inhibition from iNOS by consuming NO during steady-state catalysis, enhancing iNOS catalytic activity and NO production. MPO also inactivates eNOS through HOCl, inducing uncoupling, and reacts with L-arginine, inhibiting NO synthesis. HOCl-induced methylation of L-arginine contributes to inflammatory cardiovascular disease. Peroxynitrite, another reactive species, further promotes nitration of tyrosine residues and initiates lipid peroxidation [20].
The consequence of MPO activity at inflammatory sites is the consumption of NO, the generation of a series of reactive oxidant species responsible for inducing endothelial dysfunction. Oxidative stress associated with MPO activity may contribute to the development and progression of various cardiovascular diseases [21], including CAD [22], congestive heart failure [22], arterial hypertension [22], pulmonary arterial hypertension [22], peripheral arterial disease [22], myocardial ischemia [22], and venous thrombosis [22]. iNOS is strongly linked to NO regulation. The assertion of oxidative reactions in the vascular wall is known, but the direct implication of iNOS in these events or the exact molecular mechanism is not yet fully defined.
| Atherosclerosis Pathogenesis | ▴Top |
The concepts of atherosclerosis pathogenesis are based on the spatial heterogeneity of atherosclerosis, i.e., the focal and diffuse formation of lesions. The monoclonal multicentric hypothesis of atherogenesis proposes that atheromas arise as benign leiomyomas of the blood vessel wall, built from a monotony of different molecular markers with clonal expansion of smooth muscle cells. Atherogenic hemodynamics, characterized by biomechanical stimuli, has proven to be crucial in the pathology of atherosclerosis, including the activation of biochemical proinflammatory agonists (interleukin-1 (IL-1), tumor necrosis factor (TNF), oxidized low-density lipoprotein (oxLDL)) and the consequent onset and progression of the disease.
Oxidative stress, endothelial dysfunction, and reduced NO bioavailability induce an inflammatory response, namely endothelial proinflammatory activation.
The oxidative modification of atherosclerosis is based on the hypothesis of LDL oxidation modification as an early event in the development of atherosclerosis [11]. Endothelial dysfunction plays a critical role in atherosclerosis progression, leading to cardiovascular complications [23].
Atherosclerosis initiates upon endothelial dysfunction accompanied by LDL retention and its modification in the intima [22, 24, 25]. Modified LDL leads to the recruitment of monocytes within the intima, which differentiate and, along with vascular smooth muscle cells (VSMCs), phagocytize oxLDL, forming foam cells [24, 26, 27], activating several inflammatory signaling pathways, and creating fatty streaks, representing the first sign of atherosclerosis [24, 28]. The research states that induction of iNOS expression by oxLDL inhibits the migration of foam cells derived from macrophages, highlighting iNOS as a potential target to reduce plaque formation and atherosclerosis. Foam cells of macrophage origin identified in the atherosclerotic lesion induce proinflammatory mediators, including cytokines and chemokines, producing large amounts of oxidative species such as superoxide anion and HOCl. The coordinated action of activated endothelial cells, smooth muscle cells, monocytes/macrophages, and lymphocytes results in the production of a cellular paracrine milieu of cytokines, growth factors, and ROS within the vascular wall, prolonging chronic proinflammatory conditions and promoting the advancement of atherosclerotic lesions [29]. The mentioned inflammatory mediators can promote inflammation in the plaque and contribute to lesion progression. A fibroatheroma with a thin cap is a type of unstable plaque characterized by a thin fibrous cap, a large lipid-rich necrotic core, a high content of inflammatory cells, and increased oxidative stress [30]. The proatherogenic phenotype, based on hyperlipidemia, systemic oxidative stress, endothelial dysfunction, proinflammatory state, and biological plaque activity, i.e., monocyte chemotactic activity, leukocyte adhesion to blood vessels, production of proinflammatory cytokines, activation of vascular endothelial cells, and extracellular MPO activity, determines plaque instability [31]. Monocytes and neutrophils are more pronounced in unstable plaques; increased plaque inflammation is associated with the infiltration, activation, and degranulation of neutrophils, including the activity of extracellular MPO, a proinflammatory and pro-oxidative enzyme. HOCl derived from MPO activates metalloproteinases and directly degrades collagen, the main component of the fibrous cap, providing a clear explanation of the mechanisms by which plaque caps thin, and MPO participates in the destabilization of coronary artery atherosclerotic plaques [32, 33]. The interplay between MPO and NO significantly influences inflammation sites. NO’s impact on MPO compound II formation, duration, and decay affects substrate peroxidation rates and MPO’s ability to perform oxidation reactions [34]. These interactions have broad implications for initiating and progressing local inflammatory and cardiovascular events in vivo, promoting endothelial dysfunction and causing impaired vasoreactivity and instability of atherosclerotic plaques. The stimulated expression of iNOS promotes the formation of peroxynitrite, resulting in reduced NO production and eNOS activity, thereby linking it to the development of coronary artery atherosclerosis [4].
Endothelial dysfunction, microvascular disease, and vasospasm may exist alone or in combination with coronary atherosclerosis. It can be the dominant cause of myocardial ischemia in some patients, indicating that the spectrum of stable ischemic heart disease, as a synonym for obstructive coronary atherosclerosis, is too simplified a view [35].
| The Role of iNOS and MPO in the Development of Atherosclerosis | ▴Top |
Inflammation is a major factor in the development of CAD and a key contributor to plaque vulnerability, rupture, and ischemic cardiomyopathy with the proatherogenic effect of the inflammatory markers MPO and iNOS correlating with their concentration [36].
Clinical studies show that MPO concentrations correlate with endothelial dysfunction and the severity of CAD (coronary stenosis, coronary thrombosis, plaque ulceration) [37, 38]. Elevated concentrations of MPO in plasma have been recorded in patients with CAD, unstable angina, and acute myocardial infarction [39-42]. MPO operates in all stages of atherosclerosis. Excessive pro-oxidative activity of MPO reduces the bioavailability of NO, inhibiting the activity of eNOS, generating superoxides in the endothelium, and leading to the progression of atherosclerosis. MPO participates in atherosclerotic events such as endothelial dysfunction, destabilization of atherosclerotic plaques, and oxidation of lipoproteins [41]. By producing HOCl, MPO is crucial in plaque destabilization, so its activity is more intense in less stable plaques [37, 43-44]. The role of eNOS in the development of atherosclerosis is highly pronounced. Previous studies have reported reduced eNOS expression in severe atherosclerotic lesions in coronary arteries [45-49]. Research indicates that NO can bind to MPO in both its ferric and ferrous states, creating stable nitrosyl complexes [20, 50]. However, NO’s binding to the ferrous form of MPO is significantly slower than to the ferric form, suggesting that heme reduction affects NO’s affinity for the enzyme [20, 51]. Another aspect of MPO and NO interaction involves MPO consuming NO produced by iNOS during its catalysis, preventing the inhibition of iNOS by NO.
This process enhances iNOS’s catalytic activity and its production of citrulline and NO. MPO thus serves as a NO sink, facilitating continuous NO production by iNOS. MPO-H2O2 reaction pathways may play a significant role in increasing the catalytic activity of iNOS [52]. In inflammatory atherosclerotic diseases of coronary blood vessels, increased MPO activity and a positive correlation with inflammation enhance the catalytic activity of iNOS through a feedback mechanism, removing free NO from the iNOS milieu via MPO using H2O2 as a substrate [11, 52]. NO regulates the catalytic activity of iNOS by competing with O2 at the catalytic site of the enzyme, creating inactive NOS-nitrosyl complexes, thereby reducing the activity of iNOS and preventing the full capacity of iNOS to convert L-arginine into the product NO, which is assessed by the increased levels of citrulline, nitrate, and nitrite. The harmful effects of iNOS/NO, resulting from increased iNOS activity, contribute to the generation of ROS, peroxynitrite (ONOO-), and increased oxidative stress. Increased iNOS activity leads to the activation of the MPO-H2O2 pathway and the generation of HOCl, which causes focal vasoconstriction of the damaged coronary blood vessel. Blocking NOS with the L-arginine analog reduces NO-mediated vasodilation. The metabolic flow caused by metabolic mediators (NO, HOCl) plays a role in determining vascular tone. Considering that iNOS/NO signaling is significant in modulating the inflammatory response via a feedback mechanism, we conclude that the control of vascular tone is mediated by the MPO-H2O2 reaction pathways (Fig. 1). In the early stages of the atherosclerotic process, the regulation of NOS isoforms is unbalanced, and iNOS is significantly involved in the pathophysiology of inflammation. It is induced by T-helper cells (Th cells) and cytokines (IL-1, TNF, gamma-interferon). The cytokine-iNOS isoform is present in many tissues, including the lungs, liver, kidneys, and heart, meaning it is involved in most diseases with excessive NO production [52]. Kinetic and structural analysis of iNOS has revealed that, with increased iNOS expression in inflamed areas, the main pathway causing excessive NO production is the destabilization of the iNOS-nitrosyl complex [52]. iNOS induces metabolic changes in macrophages towards a proinflammatory phenotype. Considering that the anti-inflammatory phenotype of macrophages is characterized by low iNOS, it suggests that iNOS correlates with the degree of inflammation. In the literature, it is stated that subjects with type 2 diabetes mellitus (DM) and CAD have significantly higher iNOS activity and lower eNOS activity than subjects with type 2 DM without CAD [53, 54]. Increased iNOS expression has been observed in human myocardial infarction [55, 56]. Induction of iNOS produces excessive NO accompanied by increased ROS production, including peroxynitrite and superoxide, which can increase plaque vulnerability. The expression of iNOS was also proved to correlate positively with the severity of cardiac dysfunction and the expression of proinflammatory cytokines [57]. Endothelial dysfunction due to excessive oxLDL production leads to an imbalance in NOS activation, downregulating eNOS, facilitating iNOS activation, enhancing inflammatory processes within the vascular wall, and contributing to the progression of atherosclerosis [58-60].
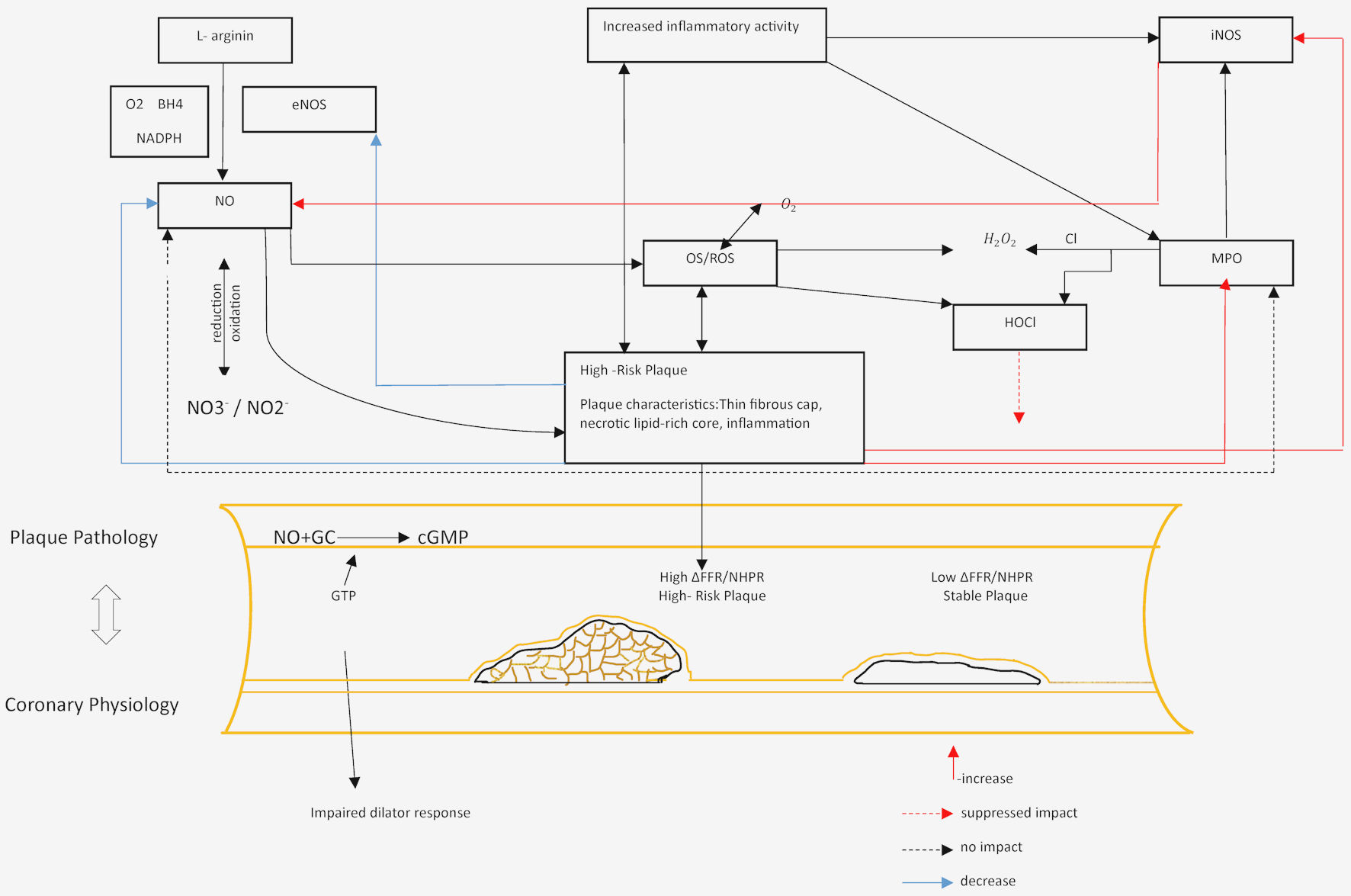 Click for large image | Figure 1. Representation of the role of iNOS in assessing the functional level of coronary artery lesions. MPO: myeloperoxidase; NO: nitric oxide; iNOS: inducible nitric oxide synthase; eNOS: endothelial nitric oxide synthase; ROS: reactive oxygen species; HOCl: hypochlorous acid; H2O2: hydrogen peroxide; NADPH: nicotinamide adenine dinucleotide phosphate; GC: guanylate cyclase; cGMP: cyclic guanosine monophosphate; FFR: fractional flow reserve; NHPR: nonhyperemic pressure ratio. |
| Interactions and Correlative Links of Functional Level of Lesions and iNOS | ▴Top |
The control of the inflammatory response develops through several molecular pathways, involving the interaction of MPO with NO, eNOS, and iNOS. The three-way relationship that exists between MPO, iNOS, and NO is a probable mechanism that governs increased NO levels at the site of inflammation. In the early stages of the atherosclerotic process in coronary blood vessels, there is an unbalanced regulation of NOS isoforms. The roles of eNOS in the development of atherosclerosis are highly pronounced. Endothelial eNOS, whose expression is limited to the vascular endothelium, becomes a source of superoxide radicals. The superoxide radical is directly associated with the modulation concentrations of NO and generates another reactive species, peroxynitrite, resulting in reduced NO production and eNOS activity [61]. The role of MPO in vascular disease involves regulating NO production through a feedback mechanism. The control of the inflammatory response through the interaction mechanism of MPO and iNOS, i.e., removing NO from the iNOS milieu via the MPO system, causes a significant increase in iNOS catalytic activity. The mutual interactions between iNOS and MPO are reflected in the pathogenesis of CCS, and their relationship is not well known. This includes the production of superoxide and other ROS through the mitochondrial electron transport chain, endoplasmic reticulum, oxidative stress, and increased flux through the hexosamine pathway. Through a feedback mechanism, MPO prevents the inhibition of iNOS by removing NO, thus preventing the formation of iNOS-nitrosyl complexes. Therefore, an unbalanced relationship between NO and MPO can modulate the catalytic activity and function of iNOS [53]. Understanding how iNOS-NO binding regulates the enzyme’s catalytic activity, the crosslinking of iNOS and MPO depending on the time, the diffusion rate of NO, local NO breakdown chemistry, and inflammatory environment (positive correlation with cytokines) can provide a better insight into the complex dual role of iNOS/NO. In conclusion, the relationship between MPO and iNOS can help in the clinical management of CCS pathophysiology. Arginine is a substrate for eNOS and the main precursor of NO in vascular endothelium. Numerous studies show that methylation of L-arginine induced by HOCl and dysfunction of the L-arginine-eNOS pathway may be involved in the pathogenesis of CAD. Therefore, the mechanisms involving ROS are associated with the pathophysiology of CAD, i.e., inducing endothelial dysfunction, leading to the progression of chronic proinflammatory processes and atherosclerotic lesions of coronary blood vessels. The role of MPO is the regulation of NO production through a feedback mechanism. The superoxide radical is directly associated with modulating NO concentration by generating another reactive species, peroxynitrite, resulting in reduced NO production and eNOS activity [20].
A number of research studies implicated that specific interactions between iNOS and MPO at sites of inflammation have been infrequently explored [52]. One mechanistic pathway shows that MPO enhances the catalytic activity of iNOS through a feedback mechanism. Their relationship, i.e., the effect of the catalytic amount of H2O2 and MPO on the iNOS-ferri-nitrosyl complex, could be a leading pathogenic factor in unveiling the phenotype and subtype of CCS.
| Functional Phenotypes of Coronary Atherosclerosis | ▴Top |
Coronary atherosclerosis can manifest as a wide range of plaque phenotypes. Phenotypes of atherosclerotic plaques determined by focal and diffuse CAD are associated with intracoronary hemodynamics [62]. Focal CAD (lipid-rich plaques and fibroatheromas with a thin cap) indicates a large pressure gradient and is a predictor of plaque rupture and unfavorable clinical outcomes. Diffuse stable CAD is characterized by calcifications and gradual changes in the pressure gradient.
Based on the cited literature, it can be concluded that NO and eNOS derived from endothelium are reduced in focal CAD, while inducible NO synthase (iNOS), activated by systemic inflammation, may be increased. Patients with focal CAD may have higher iNOS activity and high-sensitivity C-reactive protein (hsCRP) level compared to patients with diffuse CAD. Therefore, current research data implicate that the phenotypes of atherosclerotic plaques, focal and diffuse CAD, could be defined by the relationship between MPO and iNOS.
Coronary occlusive disease is one of the characteristics of CCS. These are the most common lesions that can lead to restricted blood flow under conditions of increased demand. Lesions of acute coronary syndrome (ACS) may result from a thrombus formed as a consequence of plaque disruption that does not produce critical stenosis. These findings do not imply that small atheromas precede an acute event. Large lesions causing acute myocardial infarction, due to compensatory expansion of the coronary blood vessel, may not produce critical narrowing. However, high-grade stenosis is more likely to cause acute myocardial infarction than non-occlusive lesions [63].
A substantial amount of clinical data have provided a solid scientific basis that myocardial ischemia can occur without critical coronary atherosclerosis and can be explained by overlapping effects of concealed diffuse coronary atherosclerosis [63].
It is important to emphasize that the ischemic potential of the level of coronary vascular lesions is not only a result of the development of luminal stenosis but also of the morphological characteristics of plaques characterized by oxidative stress and inflammation.
A proinflammatory and inflammatory environment can lead to increased iNOS activity, producing excessive NO followed by ROS, peroxynitrite, and superoxide production, consequently leading to severe apoptosis and plaque destabilization. iNOS in macrophages/foam cells and VSMCs correlates with the degree of inflammation in coronary blood vessels, increasing the risk of plaque rupture and acute ischemic events. iNOS is expressed in advanced atheromatous plaques, and its important role is in mediating both progressive and regressive changes in these plaques [64]. Therefore, the mentioned functions of iNOS and its involvement in the pathophysiology of inflammation are significant in assessing the morphological characteristics of plaques, plaque vulnerability, as well as the functional level of lesions (Fig. 2).
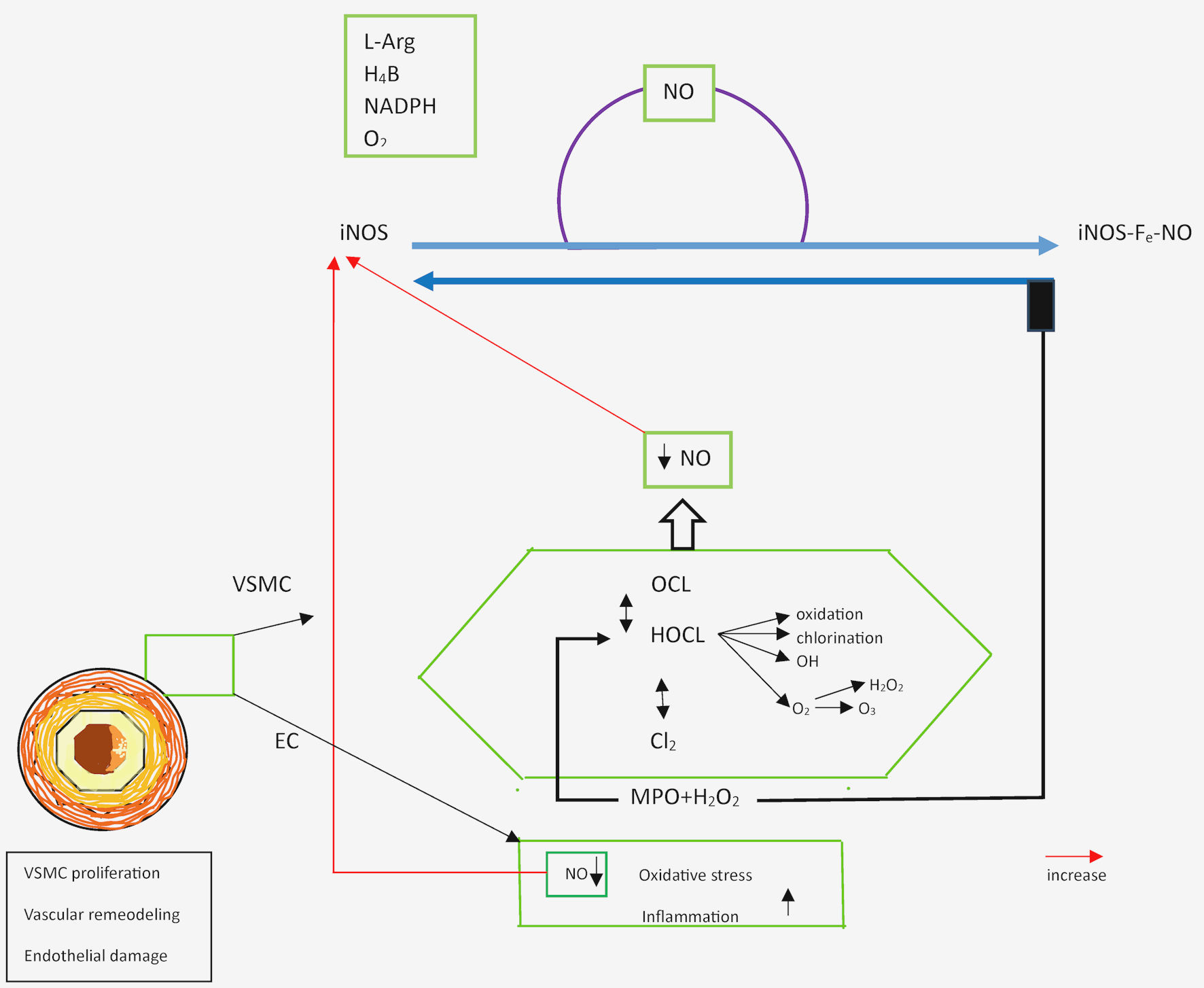 Click for large image | Figure 2. Schematic representation of the control of vascular tone mediated by the MPO-H2O2 reaction pathway. MPO: myeloperoxidase; NO: nitric oxide; iNOS: inducible nitric oxide synthase; HOCl: hypochlorous acid; H2O2: hydrogen peroxide; H4B: tetrahydrobiopterin; VSMC: vascular smooth muscle cell; NADPH: nicotinamide adenine dinucleotide phosphate; L-Arg: L-arginine; EC: endothelial cell. |
A large number of events support the clinical importance of the functional level of lesions, and currently, determining only the plaque morphology cannot reliably encompass the subset of lesions that cause severe events. Interactions between plaque morphology and local hemodynamics are still insufficiently known [63]. Some studies suggest that plaque size remains the most important diagnostic factor and is superior to plaque vulnerability [65, 66]. Plaque vulnerability has a low positive predictive value, and the benefit of revascularization based on plaque vulnerability remains unclear [65, 67]. Despite numerous methods for the pathophysiological connection of plaque composition with coronary events, there is some uncertainty about high-risk plaque as an isolated prognostic marker [65]. Certain studies link high-risk plaques to lesion severity according to different physiological indices [65, 68]. Based on the pathological mechanism of plaque rupture, most studies indicate that future severe events are associated with different plaque characteristics [65, 66, 68]. The concept of complementary roles prevails over the limitation of individual assessments.
| Conclusions | ▴Top |
If we are to improve the assessment and management of patients with CCS, a shift from a physiology-guided strategy to an anatomy-guided approach and biological activity is required. Coronary atherosclerosis manifests in diverse plaque phenotypes, each influencing clinical outcomes differently. The ischemic potential of coronary lesions arises not only from luminal stenosis but also from plaque morphology characterized by oxidative stress and inflammation. While plaque size remains a crucial diagnostic factor, the complexity of interactions between plaque characteristics and local hemodynamics necessitates a multifaceted approach to predicting severe coronary events.
MPO concentrations are closely linked to endothelial dysfunction and the severity of CAD, playing a key role in all stages of atherosclerosis by reducing NO bioavailability and inhibiting eNOS activity. MPO’s involvement in plaque destabilization and oxidation of lipoproteins highlights its significance in atherosclerotic events. Furthermore, the interaction between MPO and NO, particularly through the MPO-H2O2 pathways, modulates iNOS activity and vascular tone, emphasizing MPO’s critical role in inflammatory atherosclerotic diseases. The intricate interplay between MPO and iNOS plays a crucial role in the pathophysiology of CAD by modulating NO production and influencing inflammatory responses. Understanding the regulation of NO and the feedback mechanisms involving MPO and iNOS provides valuable insights into the progression of atherosclerosis and plaque formation. Current research suggests that the phenotypes of atherosclerotic plaques, whether focal or diffuse, may be defined by the relationship between MPO and iNOS.
In conclusion, the early stages of atherosclerosis involve unbalanced regulation of NOS isoforms, with iNOS playing a significant role in inflammation. Induced by cytokines and Th cells, iNOS correlates with the degree of inflammation and contributes to excessive NO and ROS production, increasing plaque vulnerability. The elevated expression of iNOS in conditions like type 2 DM and myocardial infarction underscores its association with severe cardiac dysfunction and the progression of atherosclerosis.
Considering the new approach to studying the functional level of coronary artery lesions through an integrative assessment of coronary anatomy, plaque quantity and quality, and physiological aspects of vascular lesions, the relationship between MPO and iNOS is a significant indicator of both diffuse and systemic nature of atheroma instability in atherogenesis. Future studies support the role and potential benefit of targeting iNOS in the assessment and management of atherosclerotic disease.
For the evaluation and treatment of patients with CAD caused by hyperlipidemia, a comparative analysis of iNOS as a metabolic enzyme and lipid parameters could be applied. iNOS is a regulator of lipid metabolism during inflammation, so when activated by systemic inflammation and associated lipid parameters, it could be an important pathogenic factor in the mechanism of accelerated atherosclerosis in patients with CAD.
Acknowledgments
None to declare.
Financial Disclosure
This work was funded by the Ministry of Science, Higher Education, and Youth of Sarajevo Canton, Bosnia Research (grant number 27-02-35-37081-10/23).
Conflict of Interest
The authors declare that they have no conflict of interest.
Author Contributions
Both authors contributed to the manuscript, wrote the text, summarized it, provided literature, and edited the final version. The authors read and approved the final manuscript.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
Abbreviations
CCS: chronic coronary syndrome; LDL low-density lipoprotein: ; MPO: myeloperoxidase; NO: nitric oxide; iNOS: inducible nitric oxide synthase; eNOS: endothelial nitric oxide synthase; CAD: coronary artery disease; 8-oxo-dG :8-oxo-2-deoxyguanosine: ; ROS: reactive oxygen species; HOCl: hypochlorous acid; H2O2: hydrogen peroxide; RNS: reactive nitrogen species; SOD: superoxide dismutase; O•-2: superoxide radical; CAT: catalase; GPx1: glutathione peroxidase 1; GSH: glutathione; 1O2: singlet oxygen; OH•-: hydroxyl radical; ONOO-: peroxynitrite species; nNOS: neuronal nitric oxide synthase; H4B: tetrahydrobiopterin; ACS: acute coronary syndrome; VSMCs: vascular smooth muscle cells
| References | ▴Top |
- Knuuti J, Wijns W, Saraste A, Capodanno D, Barbato E, Funck-Brentano C, Prescott E, et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur Heart J. 2020;41(3):407-477.
doi pubmed - Gallo G, Savoia C. New insights into endothelial dysfunction in cardiometabolic diseases: potential mechanisms and clinical implications. Int J Mol Sci. 2024;25(5):2973.
doi pubmed pmc - Nicholls SJ, Hazen SL. Myeloperoxidase, modified lipoproteins, and atherogenesis. J Lipid Res. 2009;50(Suppl):S346-351.
doi pubmed pmc - Batty M, Bennett MR, Yu E. The role of oxidative stress in atherosclerosis. Cells. 2022;11(23):3843.
doi pubmed pmc - Tran N, Garcia T, Aniqa M, Ali S, Ally A, Nauli SM. Endothelial Nitric Oxide Synthase (eNOS) and the cardiovascular system: in physiology and in disease states. Am J Biomed Sci Res. 2022;15(2):153-177.
pubmed pmc - Medina-Leyte DJ, Zepeda-Garcia O, Dominguez-Perez M, Gonzalez-Garrido A, Villarreal-Molina T, Jacobo-Albavera L. Endothelial dysfunction, inflammation and coronary artery disease: potential biomarkers and promising therapeutical approaches. Int J Mol Sci. 2021;22(8):3850.
doi pubmed pmc - Ighodaro OM, Akinloye OA. First-line defense antioxidants-superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPX): their fundamental role in the entire antioxidant defense grid. Alexandria Journal of Medicine. 2018;54(4):287-293.
- Lubos E, Loscalzo J, Handy DE. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2011;15(7):1957-1997.
doi pubmed pmc - Liu J, Han X, Zhang T, Tian K, Li Z, Luo F. Reactive oxygen species (ROS) scavenging biomaterials for anti-inflammatory diseases: from mechanism to therapy. J Hematol Oncol. 2023;16(1):116.
doi pubmed pmc - Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect Microbiol. 2017;7:373.
doi pubmed pmc - Frangie C, Daher J. Role of myeloperoxidase in inflammation and atherosclerosis (Review). Biomed Rep. 2022;16(6):53.
doi pubmed pmc - Gantner BN, LaFond KM, Bonini MG. Nitric oxide in cellular adaptation and disease. Redox Biol. 2020;34:101550.
doi pubmed pmc - Lowry JL, Brovkovych V, Zhang Y, Skidgel RA. Endothelial nitric-oxide synthase activation generates an inducible nitric-oxide synthase-like output of nitric oxide in inflamed endothelium. J Biol Chem. 2013;288(6):4174-4193.
doi pubmed pmc - Masha A, Dinatale S, Allasia S, Martina V. Role of the decreased nitric oxide bioavailability in the vascular complications of diabetes mellitus. Curr Pharm Biotechnol. 2011;12(9):1354-1363.
doi pubmed - Davies MJ. Myeloperoxidase-derived oxidation: mechanisms of biological damage and its prevention. J Clin Biochem Nutr. 2011;48(1):8-19.
doi pubmed pmc - Nilsson R, Liu NA. Nuclear DNA damages generated by reactive oxygen molecules (ROS) under oxidative stress and their relevance to human cancers, including ionizing radiation-induced neoplasia part I: physical, chemical and molecular biology aspects. Radiation Medicine and Protection. 2020;1(3):140-152.
- Khan AA, Alsahli MA, Rahmani AH. Myeloperoxidase as an active disease biomarker: recent biochemical and pathological perspectives. Med Sci (Basel). 2018;6(2):33.
doi pubmed pmc - Galijasevic S. The development of myeloperoxidase inhibitors. Bioorg Med Chem Lett. 2019;29(1):1-7.
doi pubmed - Shao B, Oda MN, Oram JF, Heinecke JW. Myeloperoxidase: an oxidative pathway for generating dysfunctional high-density lipoprotein. Chem Res Toxicol. 2010;23(3):447-454.
doi pubmed pmc - Galijasevic S. Myeloperoxidase interactions with nitric oxide: a review of mechanistic pathways. Bulletin of the Chem. and Techn. of BH. 2013;40:1-8.
- Ndrepepa G. Myeloperoxidase - A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin Chim Acta. 2019;493:36-51.
doi pubmed - Mundi S, Massaro M, Scoditti E, Carluccio MA, van Hinsbergh VWM, Iruela-Arispe ML, De Caterina R. Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review. Cardiovasc Res. 2018;114(1):35-52.
doi pubmed pmc - Maruhashi T, Higashi Y. Pathophysiological association between diabetes mellitus and endothelial dysfunction. Antioxidants (Basel). 2021;10(8):1306.
doi pubmed pmc - Jebari-Benslaiman S, Galicia-Garcia U, Larrea-Sebal A, Olaetxea JR, Alloza I, Vandenbroeck K, Benito-Vicente A, et al. Pathophysiology of atherosclerosis. Int J Mol Sci. 2022;23(6):3346.
doi pubmed pmc - Hermida N, Balligand JL. Low-density lipoprotein-cholesterol-induced endothelial dysfunction and oxidative stress: the role of statins. Antioxid Redox Signal. 2014;20(8):1216-1237.
doi pubmed - Yu XH, Fu YC, Zhang DW, Yin K, Tang CK. Foam cells in atherosclerosis. Clin Chim Acta. 2013;424:245-252.
doi pubmed - Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129(15):1551-1559.
doi pubmed - Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W, Jr., Rosenfeld ME, Schaffer SA, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, American Heart Association. Circulation. 1994;89(5):2462-2478.
doi pubmed - Gimbrone MA, Jr., Garcia-Cardena G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. 2016;118(4):620-636.
doi pubmed pmc - Puylaert P, Zurek M, Rayner KJ, De Meyer GRY, Martinet W. Regulated necrosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2022;42(11):1283-1306.
doi pubmed - Song B, Bie Y, Feng H, Xie B, Liu M, Zhao F. Inflammatory factors driving atherosclerotic plaque progression new insights. J Transl Int Med. 2022;10(1):36-47.
doi pubmed pmc - Hawkins CL, Davies MJ. Role of myeloperoxidase and oxidant formation in the extracellular environment in inflammation-induced tissue damage. Free Radic Biol Med. 2021;172:633-651.
doi pubmed - Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J Biol Chem. 2001;276(44):41279-41287.
doi pubmed - Galijasevic S, Saed GM, Hazen SL, Abu-Soud HM. Myeloperoxidase metabolizes thiocyanate in a reaction driven by nitric oxide. Biochemistry. 2006;45(4):1255-1262.
doi pubmed - Gutierrez E, Flammer AJ, Lerman LO, Elizaga J, Lerman A, Fernandez-Aviles F. Endothelial dysfunction over the course of coronary artery disease. Eur Heart J. 2013;34(41):3175-3181.
doi pubmed pmc - Arvunescu AM, Ionescu RF, Cretoiu SM, Dumitrescu SI, Zaharia O, Nanea IT. Inflammation in heart failure-future perspectives. J Clin Med. 2023;12(24):7738.
doi pubmed pmc - Teng N, Maghzal GJ, Talib J, Rashid I, Lau AK, Stocker R. The roles of myeloperoxidase in coronary artery disease and its potential implication in plaque rupture. Redox Rep. 2017;22(2):51-73.
doi pubmed pmc - Baseri M, Heidari R, Mahaki B, Hajizadeh Y, Momenizadeh A, Sadeghi M. Myeloperoxidase levels predicts angiographic severity of coronary artery disease in patients with chronic stable angina. Adv Biomed Res. 2014;3:139.
doi pubmed pmc - Mayyas FA, Al-Jarrah MI, Ibrahim KS, Alzoubi KH. Level and significance of plasma myeloperoxidase and the neutrophil to lymphocyte ratio in patients with coronary artery disease. Exp Ther Med. 2014;8(6):1951-1957.
doi pubmed pmc - Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286(17):2136-2142.
doi pubmed - Tangeten C, Zouaoui Boudjeltia K, Delporte C, Van Antwerpen P, Korpak K. Unexpected role of MPO-Oxidized LDLs in atherosclerosis: in between inflammation and its resolution. Antioxidants (Basel). 2022;11(5):874.
doi pubmed pmc - Mocatta TJ, Pilbrow AP, Cameron VA, Senthilmohan R, Frampton CM, Richards AM, Winterbourn CC. Plasma concentrations of myeloperoxidase predict mortality after myocardial infarction. J Am Coll Cardiol. 2007;49(20):1993-2000.
doi pubmed - Rashid I, Maghzal GJ, Chen YC, Cheng D, Talib J, Newington D, Ren M, et al. Myeloperoxidase is a potential molecular imaging and therapeutic target for the identification and stabilization of high-risk atherosclerotic plaque. Eur Heart J. 2018;39(35):3301-3310.
doi pubmed - Chen W, Tumanov S, Stanley CP, Kong SMY, Nadel J, Vigder N, Newington DL, et al. Destabilization of atherosclerotic plaque by bilirubin deficiency. Circ Res. 2023;132(7):812-827.
doi pubmed - Wilcox JN, Subramanian RR, Sundell CL, Tracey WR, Pollock JS, Harrison DG, Marsden PA. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc Biol. 1997;17(11):2479-2488.
doi pubmed - Daiber A, Xia N, Steven S, Oelze M, Hanf A, Kroller-Schon S, Munzel T, et al. New therapeutic implications of endothelial Nitric Oxide Synthase (eNOS) function/dysfunction in cardiovascular disease. Int J Mol Sci. 2019;20(1):187.
doi pubmed pmc - Venugopal SK, Devaraj S, Yuhanna I, Shaul P, Jialal I. Demonstration that C-reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106(12):1439-1441.
doi pubmed - Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104(22):2673-2678.
doi pubmed - Oemar BS, Tschudi MR, Godoy N, Brovkovich V, Malinski T, Luscher TF. Reduced endothelial nitric oxide synthase expression and production in human atherosclerosis. Circulation. 1998;97(25):2494-2498.
doi pubmed - Forbes LV, Sjogren T, Auchere F, Jenkins DW, Thong B, Laughton D, Hemsley P, et al. Potent reversible inhibition of myeloperoxidase by aromatic hydroxamates. J Biol Chem. 2013;288(51):36636-36647.
doi pubmed pmc - Abu-Soud HM, Hazen SL. Nitric oxide modulates the catalytic activity of myeloperoxidase. J Biol Chem. 2000;275(8):5425-5430.
doi pubmed - Galijasevic S, Saed GM, Diamond MP, Abu-Soud HM. Myeloperoxidase up-regulates the catalytic activity of inducible nitric oxide synthase by preventing nitric oxide feedback inhibition. Proc Natl Acad Sci U S A. 2003;100(25):14766-14771.
doi pubmed pmc - Habib SS, Al-Regaiey KA, Al-Khlaiwi T, Habib SM, Bashir S, Al-Hussain F, Habib SH. Serum inducible and endothelial nitric oxide synthase in coronary artery disease patients with Type 2 Diabetes mellitus. Eur Rev Med Pharmacol Sci. 2022;26(10):3695-3702.
doi pubmed - Adela R, Nethi SK, Bagul PK, Barui AK, Mattapally S, Kuncha M, Patra CR, et al. Hyperglycaemia enhances nitric oxide production in diabetes: a study from South Indian patients. PLoS One. 2015;10(4):e0125270.
doi pubmed pmc - Lind M, Hayes A, Caprnda M, Petrovic D, Rodrigo L, Kruzliak P, Zulli A. Inducible nitric oxide synthase: Good or bad? Biomed Pharmacother. 2017;93:370-375.
doi pubmed - Wilmes V, Scheiper S, Roehr W, Niess C, Kippenberger S, Steinhorst K, Verhoff MA, et al. Increased inducible nitric oxide synthase (iNOS) expression in human myocardial infarction. Int J Legal Med. 2020;134(2):575-581.
doi pubmed - Yu X, Ge L, Niu L, Lian X, Ma H, Pang L. The dual role of inducible nitric oxide synthase in myocardial ischemia/reperfusion injury: friend or foe? Oxid Med Cell Longev. 2018;2018:8364848.
doi pubmed pmc - Gliozzi M, Scicchitano M, Bosco F, Musolino V, Carresi C, Scarano F, Maiuolo J, et al. Modulation of nitric oxide synthases by oxidized LDLs: role in vascular inflammation and atherosclerosis development. Int J Mol Sci. 2019;20(13):3294.
doi pubmed pmc - Colasanti M, Suzuki H. The dual personality of NO. Trends Pharmacol Sci. 2000;21(7):249-252.
doi pubmed - Stancu CS, Toma L, Sima AV. Dual role of lipoproteins in endothelial cell dysfunction in atherosclerosis. Cell Tissue Res. 2012;349(2):433-446.
doi pubmed - Bil P, Ciesielska S, Jaksik R, Rzeszowska-Wolny J. Circuits regulating superoxide and nitric oxide production and neutralization in different cell types: expression of participating genes and changes induced by ionizing radiation. Antioxidants (Basel). 2020;9(8):701.
doi pubmed pmc - Sakai K, Mizukami T, Leipsic J, Belmonte M, Sonck J, Norgaard BL, Otake H, et al. Coronary atherosclerosis phenotypes in focal and diffuse disease. JACC Cardiovasc Imaging. 2023;16(11):1452-1464.
doi pubmed - Libby P, Bonow RO, Mann DL, Tomaselli GF, Bhatt D, Solomon SD, Braunwald E. Heart failure with preserved and mildly reduced ejection fraction. In: Lam CSP, Shah SJ, Solomon SD. Braunwald’s Heart Disease. 12 th ed. USA: Elsevier. 2022; pp.425-441.
- Khattab AD, Ali IS, Dils R, Kerr D, Jenkinson DF, Allen S, Rana M. Nitric oxide synthase and carotid artery plaque morphology. Atherosclerosis Supplements. 2001;2(2):103.
- Yang S, Koo BK, Narula J. Interactions between morphological plaque characteristics and coronary physiology: from pathophysiological basis to clinical implications. JACC Cardiovasc Imaging. 2022;15(6):1139-1151.
doi pubmed - Arbab-Zadeh A. Does "Vulnerable" atherosclerotic plaque modify coronary blood flow? How myths perpetuate. JACC Cardiovasc Imaging. 2020;13(3):757-759.
doi pubmed - Kaul S, Narula J. In search of the vulnerable plaque: is there any light at the end of the catheter? J Am Coll Cardiol. 2014;64(23):2519-2524.
doi pubmed - Panagiotis X, Quentin de H. The relationship between plaque morphology and its hemodynamic significance. American College of Cardiology. 2018.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.