

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website https://www.cardiologyres.org |
Original Article
Volume 15, Number 5, October 2024, pages 404-414

Fibroblast Growth Factor 21 Suppressed Neutrophil Extracellular Traps Induced by Myocardial Ischemia/Reperfusion Injury via Adenosine Monophosphate-Activated Protein Kinase
Ling Yun Gua, Cheng Gao Jiab, Zuo Zhen Shenga, Wen Long Jianga, Zhuo Wen Xub, Wei Zhang Lib, Jun You Cuia, Hua Zhanga, b, c
aDepartment of Cardiology, Jiangyin Hospital Affiliated to Nantong University, Jiangyin, China
bThe Jiangyin Clinical College of Xuzhou Medical University, Jiangyin, China
cCorresponding Author: Hua Zhang, The Jiangyin Clinical College of Xuzhou Medical University, Jiangyin, China
Manuscript submitted July 24, 2024, accepted September 6, 2024, published online October 11, 2024
Short title: FGF21 Inhibited NETs Expression via AMPK
doi: https://doi.org/10.14740/cr1705
| Abstract | ▴Top |
Background: Previous investigations have established the anti-inflammatory properties of fibroblast growth factor 21 (FGF21). However, the specific mechanism through which FGF21 mitigates myocardial ischemia/reperfusion (I/R) injury by inhibiting neutrophil extracellular traps (NETs) remains unclear.
Methods: A mice model of myocardial I/R injury was induced, and myocardial tissue was stained with immunofluorescence to assess NETs. Serum NETs levels were quantified using a PicoGreen kit. In addition, the expression levels of adenosine monophosphate (AMP)-activated protein kinase (AMPK) and FGF21 were evaluated by Wes fully automated protein blotting quantitative analysis system. Moreover, a hypoxia/reoxygenation (H/R) model was established using AMPK inhibitor and agonist pretreated H9c2 cells to further explore the relationship between FGF21 and AMPK.
Results: Compared with the control group, serum NETs levels were significantly higher in I/R mice, and a large number of NETs were formed in myocardial tissues (97.63 ± 11.45 vs. 69.65 ± 3.33, P < 0.05). However, NETs levels were reversed in FGF21 pretreated mice (P < 0.05). Further studies showed that FGF21 enhanced AMPK expression, which was significantly increased after inhibition of AMPK and decreased after promotion of AMPK (P < 0.05).
Conclusions: FGF21 may exert cardioprotective effects by inhibiting I/R injury-induced NETs via AMPK.
Keywords: Fibroblast growth factor 21; Myocardial ischemia/reperfusion injury; Neutrophil extracellular traps; AMP-activated protein kinase
| Introduction | ▴Top |
Acute myocardial infarction (AMI) is a common clinical cardiovascular emergency. Emergency percutaneous coronary intervention (PCI), which significantly reduces mortality, is the main strategy for reperfusion therapy in patients with AMI [1]. However, myocardial reperfusion therapy, while essential for restoring coronary blood flow, may also cause additional myocardial damage known as myocardial ischemia/reperfusion (I/R) injury, which accounts for approximately 50% of the final myocardial area of injury [2-4]. Despite technological advances in myocardial reperfusion therapy, including continuous improvements in PCI techniques and drugs, an effective approach for preventing I/R injury remains elusive [5].
Myocardial I/R injury involves various pathophysiological mechanisms, prominently featuring the inflammatory response, oxidative stress, and mitochondrial dysfunction. Among these factors, the inflammatory response and oxidative stress are pivotal [6]. In the initial hours of AMI, inflammatory cells, notably neutrophils, extensively infiltrate the infarcted area [7]. Neutrophils become activated in response to various factors, including proinflammatory cytokines, activated platelets, damaged vascular endothelium, and oxygen free radicals. Activated neutrophils release chromatin and granulocyte enzymes outside the cell, forming a net-like structure termed neutrophil extracellular traps (NETs) [8]. While NETs play a critical role in the inflammatory response and benefit various infectious diseases, they promote inflammation in sterile diseases by releasing active molecules in substantial quantities [9].
NET levels in the peripheral blood of AMI patients are correlated with disease severity and prognosis, and elevated NET levels in intracoronary thrombi are correlated with a reduced left ventricular ejection fraction and infarct size [10-13]. Additionally, NET levels independently predict major adverse cardiac event (MACE) occurrence within a year following AMI [14]. NETs promote thrombosis by causing damage to vascular endothelial cells, inducing an inflammatory response, serving as a scaffold for platelet aggregation and activation, and activating the coagulation system while inhibiting the fibrinolytic process [15]. In a mouse AMI model, the anti-inflammatory effects of colchicine significantly inhibited NET formation and inflammatory responses, improved myocardial tissue remodeling, and increased survival rates [16].
Fibroblast growth factor 21 (FGF21), which has anti-inflammatory properties and attenuates myocardial I/R injury, is significantly increased in the serum of patients with AMI and is correlated with MACE [17, 18]. However, whether FGF21 can mitigate myocardial I/R injury by inhibiting NETs remains unexplored. In this study, we explored the mechanism of FGF21 inhibition of NETs during I/R injury by using a mouse I/R injury model and a myocardial hypoxia/reoxygenation (H/R) model.
| Materials and Methods | ▴Top |
Experimental study design
Six- to eight-week-old male C57BL/6WT mice were purchased from Spivey (Beijing) Biotechnology Co. All the mice were maintained in specific-pathogen-free (SPF) chambers at temperatures between 20 and 26 °C, humidity between 40% and 75%, and a light/dark cycle of 12 h. The mice had free access to standard laboratory chow and water. Following 1 week of adaptive feeding, the mice were randomly allocated to one of the four groups: the control group (n = 6), I/R group (n = 6), I/R + FGF21 group (n = 6), and I/R + compound C group (n = 6). All protocols in this study were approved by the Nantong University Laboratory Animal Center (approval no. S20210820-002) in accordance with all the applicable institutional ethical guidelines for the care, welfare and use of animals.
Before the myocardial I/R injury model was established, the mice in the I/R + FGF21 group received subcutaneous injections of recombinant FGF21 protein (0.1 mg/kg/day, Novoprotein, China) for 14 consecutive days. The mice in the I/R + compound C group were intraperitoneally injected with 5 mg/kg compound C (MCE, USA) 1 h before I/R injury was induced.
H9c2 cells were pretreated with compound C (10 µmol/L, MCE, USA) and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) (1 mmol/L, MCE, USA) for 24 h before modeling. H9c2 cells were then randomly divided into a control group (n = 3), H/R group (n = 3), H/R + compound C group (n = 3), and H/R + AICAR group (n = 3).
Establishment of an animal model of myocardial I/R injury
After the mice were subjected to 10% chloral hydrate (3.5 mL/kg) intraperitoneal anesthesia, the fourth rib at the left margin of the sternum was clipped. After the pericardium was bluntly separated and clipped, the anterior descending branch was ligated with sutures under a body mirror to establish a model of myocardial I/R injury. The I/R injury model was induced by occluding the left anterior descending coronary artery for 30 min, followed by 24 h of reperfusion. There were no extra surviving mice in this experiment due to the strict control of the use of experimental animals. The mice were sacrificed by cervical dislocation at the end of the experiment. The hearts were promptly immersed in liquid nitrogen for preservation. Peripheral blood samples were collected, and after centrifugation at 3,000 rpm for 10 min, the supernatant was stored in tubes at -80 °C.
Cell culture and establishment of H/R models
H9c2 cardiomyocytes obtained from ATCC were cultured in a medium comprising 90% Dulbecco’s modified Eagle medium (DMEM) and 10% fetal bovine serum (FBS) and maintained in an incubator at 37 °C with 5% CO2. To establish anaerobic conditions, H9c2 cardiomyocytes were placed in a 2.5 L sealed culture jar (MGC, Japan) with a 2.5 L anaerobic gas-producing bag (MGC, Japan). The sealed jars were incubated at 37 °C for 12 h. H9c2 cells were subsequently removed from the sealed jars and incubated for an additional 24 h at 37 °C to establish the H/R model.
2,3,5-Triphenyl tetrazolium chloride (TTC) staining
The myocardial tissue was rapidly cooled to -20 °C for approximately 15 min. Subsequently, it was meticulously sliced into five 2 mm thick sections in the coronal direction. These sections were then immersed in a 2% TTC solution at 37 °C until normal myocardial tissue displayed a red hue, while the infarcted tissue exhibited a white appearance. Once a color change was achieved, the sections were carefully retrieved. The treated sections were immersed in a 4% paraformaldehyde solution for 1 h and subsequently examined at the macro level. Using Image-Pro Plus 5.0 image analysis software, we quantified the area of myocardial infarction. To calculate the volume of myocardial infarction for each section, the mean number of infarction areas was multiplied by the section thickness. The total myocardial infarct volume of the mice was then determined by summing the infarct volumes of all the slices. To express the magnitude of myocardial infarction as a percentage, the infarct volume was divided by the total volume, and the resulting value was multiplied by 100%.
Hematoxylin and eosin (H&E) staining
The myocardial tissue was fixed with a 4% formaldehyde solution and dehydrated through a series of graded ethanol solutions (75%, 85%, 95%, 95%, 100%, and 100%). After being washed with xylene, the myocardial tissues were embedded in paraffin and sectioned at a thickness of 2 µm. The resulting sections were subjected to a sequential treatment involving immersion in xylene, graded ethanol, pure water, and hematoxylin staining solution. Next, the sections were rinsed with tap water and reblued with Scott’s bluing solution. To facilitate staining, the rinsed sections were then immersed in eosin staining solution, followed by swift dehydration in a graded ethanol series and sealing with a sealer. The sections were meticulously examined using an Olympus microscope at various angles and magnifications, and photographs were captured for subsequent analysis. The obtained results were subjected to detailed analysis.
Immunofluorescence staining
The dewaxed sections were subjected to heat treatment in an autoclave with an antigen repair solution until boiling. After natural cooling, the sections were subjected to treatment with 5% bovine serum albumin (Solarbio, China) for 30 min at 37 °C. Primary antibodies, specifically myeloperoxidase (MPO) (Proteintech, USA) and histone H3 (Abcam, UK), were used to incubate the myocardial tissue sections overnight at 4 °C. Following this incubation, the sections were rinsed with phosphate-buffered saline (PBS) and then treated with either Cy3-conjugated goat anti-rabbit IgG antibody (ABdonal, China) or goat anti-rabbit IgG/488 (ZSGB, China) for 30 min at 37 °C in the dark. Subsequently, the nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) (KeyGEN, China), and the sections were blocked with a solution containing an anti-fluorescence quencher. The treated sections were then observed and analyzed via a fluorescence microscope (BX53, Olympus). The percentage of myocardial infarction volume was calculated as follows: myocardial infarction volume (%) = the sum of the infarct volumes of all slices/the left ventricular volume × 100%.
PicoGreen quantification of dsDNA
The working solution was prepared following the instructions provided with the PicoGreen kit (Yesen, China). Subsequently, 100 µL of the sample and 100 µL of the prepared working solution were added to a 96-well plate and incubated at room temperature for 5 min while avoiding exposure to light. The fluorescence intensity of the samples was measured via an enzyme marker with excitation at 480 nm and emission at 520 nm.
Cell activity assay
The medium from H9c2 cardiomyocytes inoculated into 96-well plates was aspirated. Subsequently, 80 µL of CellCounting-Lite 2.0 (Vazyme, China) was added to each well, shaken, mixed, and left at room temperature for 5 min. Following this incubation, the luminescence signal was detected.
Using the Wes Fully Automated Protein Blotting and Quantitative Analysis System for detecting protein expression
Proteins were extracted from mouse myocardial tissue and H9c2 cells via lysis and centrifugation, and their concentrations were determined via the BCA method (MA0082, China). For the samples, primary antibodies, including FGF21 (Abcam, UK), adenosine monophosphate (AMP)-activated protein kinase (AMPK) (Affinity, USA), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (TransGen, China), and luminescent solutions, were prepared according to the guidance instructions of the Wes Fully Automated Protein Blotting and Quantitative Analysis System (Bio-Techne, USA), after which they were added sequentially to the plates and run on the machine. The assay results were analyzed via Compass software.
Statistical analysis
Statistical analyses were conducted using the SPSS 22.0 statistical package, and graphical representations were generated with GraphPad Prism 8.0.0. Quantitative variables are presented as the means ± standard deviations and were compared using the Student’s t-test. For comparisons involving multiple groups, one-way analysis of variance (ANOVA) was employed, and statistical significance was considered at a P value of < 0.05 (two-tailed).
| Results | ▴Top |
FGF21 reduced the volume of myocardial I/R injury
The myocardial infarction volume post-I/R was assessed via TTC staining. The results revealed that, compared with the control mice, the mice subjected to I/R injury developed larger myocardial infarcts, constituting approximately 11.15±2.97% of the infarct volume in the left ventricle (P < 0.05). In contrast, the percentage of myocardial infarct volume in mice pretreated with FGF21 before I/R injury was approximately 7.69±1.75%, which was significantly lower than that in the I/R group (P < 0.05) (Fig. 1). However, there was no significant difference in the myocardial infarction volume between the control and control + FGF21 groups. These results showed that FGF21 could reduce the infarct volume caused by myocardial I/R injury.
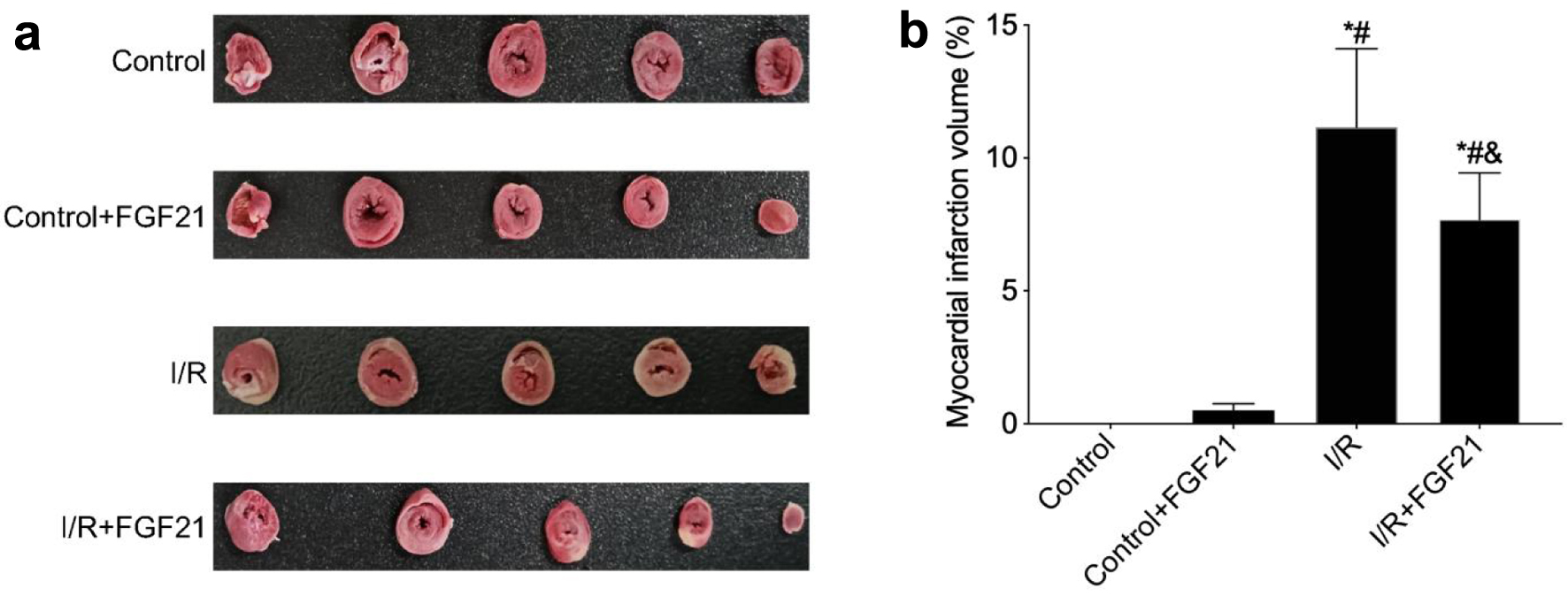 Click for large image | Figure 1. FGF21 inhibited I/R injury in myocardial tissue (TTC staining). (a) TTC staining of myocardial tissue. (b) Myocardial infarction volume calculated based on TTC staining. *P < 0.05 versus control group; #P < 0.05 versus control + FGF21 group; &P < 0.05 versus I/R group. FGF21: fibroblast growth factor 21; I/R: ischemia/reperfusion; TTC: 2,3,5-triphenyl tetrazolium chloride. |
FGF21 attenuated myocardial tissue damage caused by I/R injury
H&E staining revealed that the myocardial cells in the control group and the control + FGF21 group presented a neat and tightly arranged structure, a regular morphology, and no apparent edema or inflammatory cell infiltration. Conversely, myocardial cells in the I/R injury group exhibited abnormal morphology, disorganized arrangement, and a substantial influx of inflammatory cells. Notably, pathological changes in the myocardium were markedly improved in mice with I/R injury pretreated with FGF21 (Fig. 2). These findings demonstrated the ability of FGF21 to attenuate myocardial I/R injury.
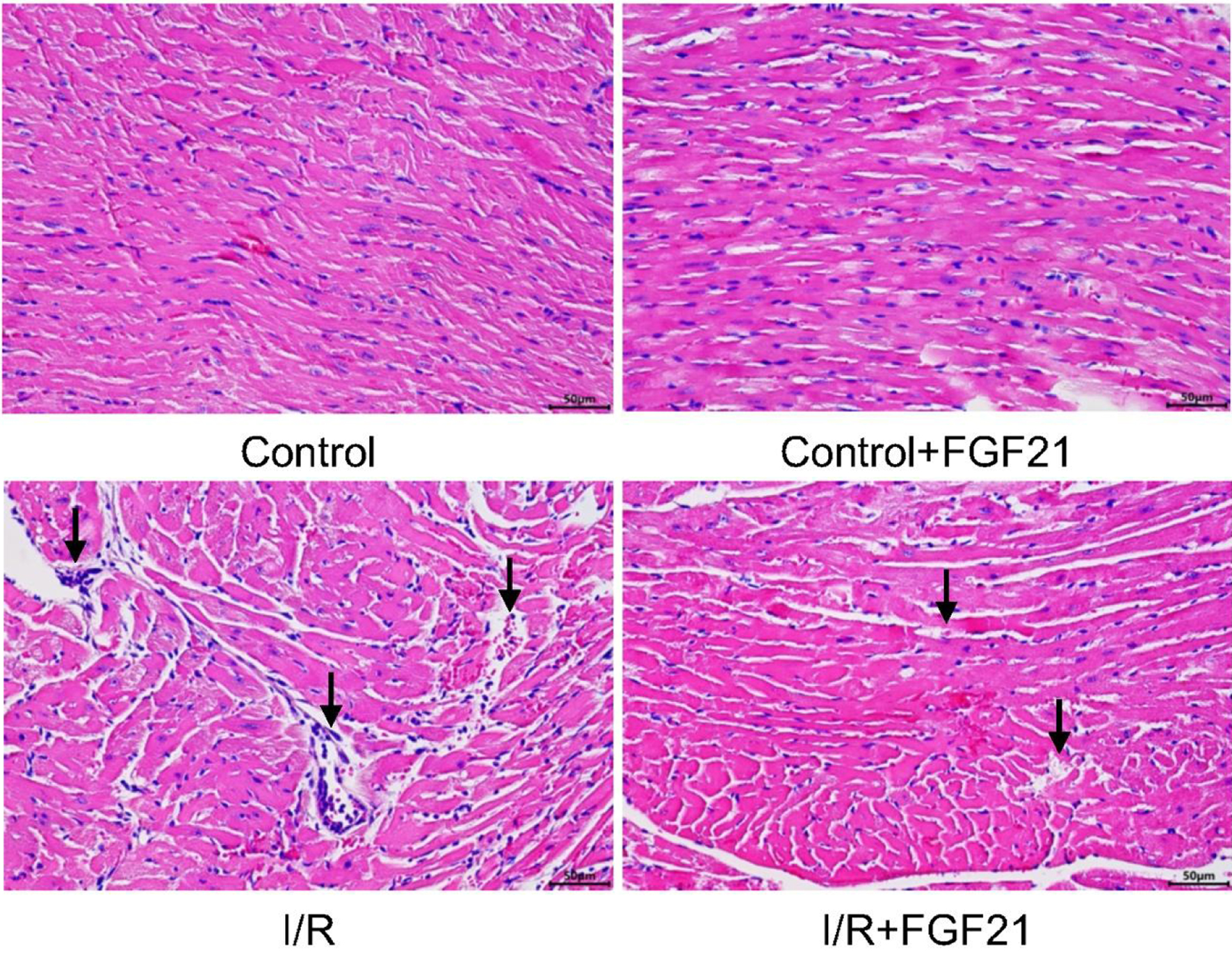 Click for large image | Figure 2. Myocardial tissue after myocardial I/R injury in mice pretreated with FGF21 (H&E staining, × 400). The black arrows represent obvious pathological changes in myocardial tissue. FGF21: fibroblast growth factor 21; H&E: hematoxylin and eosin; I/R: ischemia/reperfusion. |
FGF21 inhibited I/R injury-induced NETs
To further investigate the mechanism by which FGF21 attenuates myocardial I/R injury, we first examined NET expression after myocardial I/R injury via immunofluorescence. Immunofluorescence revealed almost no NET formation in the control group or the control + FGF21 group. However, histone H3 and MPO expression significantly increased in the myocardial tissue of the I/R group, indicating substantial NET expression due to the presence of I/R (P < 0.05). In contrast, NET expression was significantly lower in I/R-injured mice pretreated with FGF21 compared to those in the I/R group (P < 0.05) (Fig. 3).
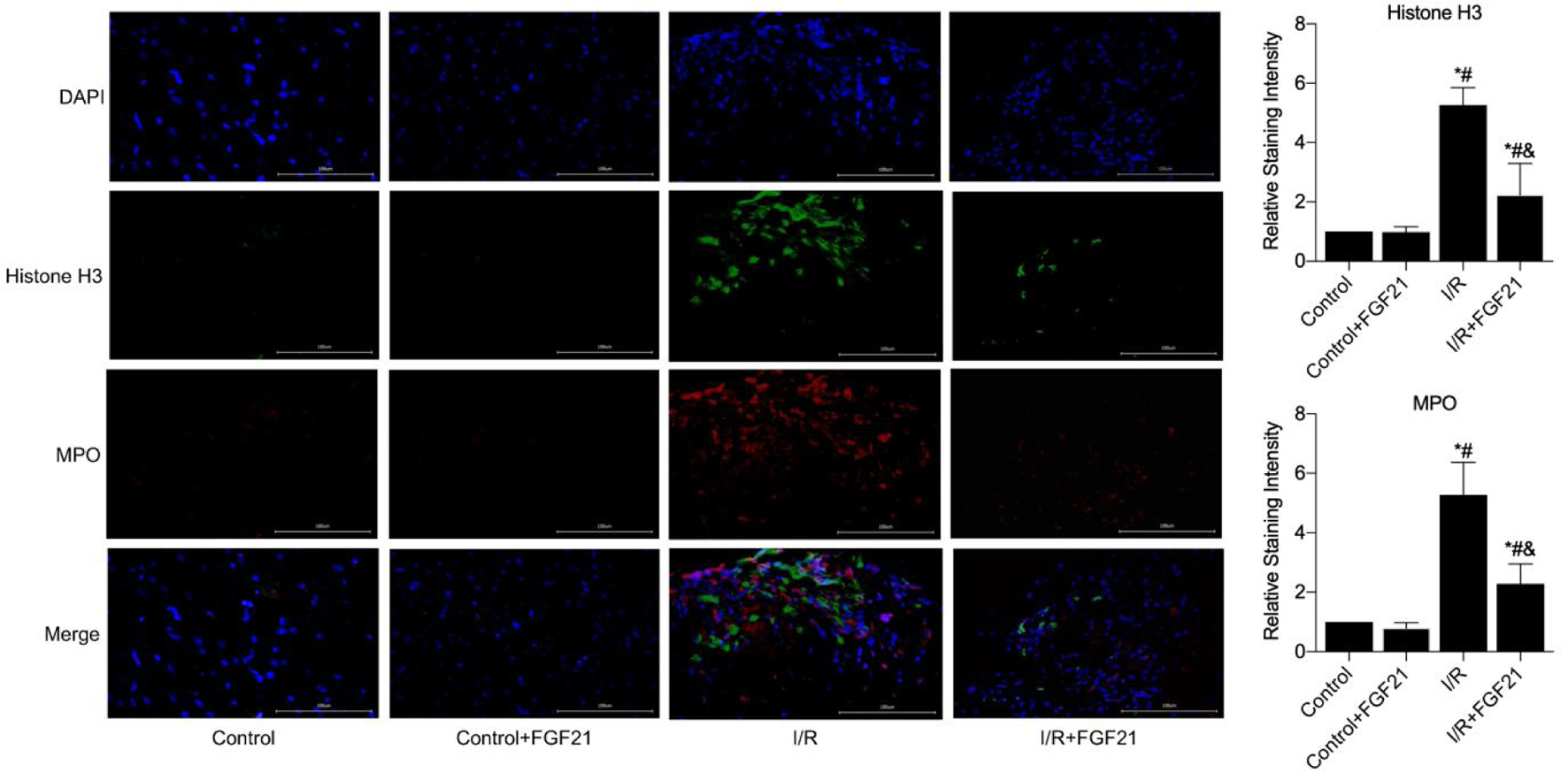 Click for large image | Figure 3. NETs formation in myocardial necrotic tissues was detected after the establishment of a myocardial I/R injury model in FGF21 pretreated mice (immunofluorescence, × 400). *P < 0.05 versus control group; #P < 0.05 versus control + FGF21 group; &P < 0.05 versus I/R group. FGF21: fibroblast growth factor 21; I/R: ischemia/reperfusion; NETs: neutrophil extracellular traps. |
Using PicoGreen, the detection of dsDNA, the main component of NETs, revealed that the serum levels of NETs were significantly greater in the I/R group (97.63 ± 11.45 vs. 69.65 ± 3.33) compared to the control group (P < 0.05). In contrast, NET levels were significantly lower in the I/R + FGF21 group compared to the I/R group (69.60 ± 5.66 vs. 97.63 ± 11.45, P < 0.05) (Fig. 4). However, there was no statistically significant difference in NET levels between the control and control + FGF21 groups. These studies demonstrated that FGF21 could inhibit the expression of NETs induced by myocardial I/R injury.
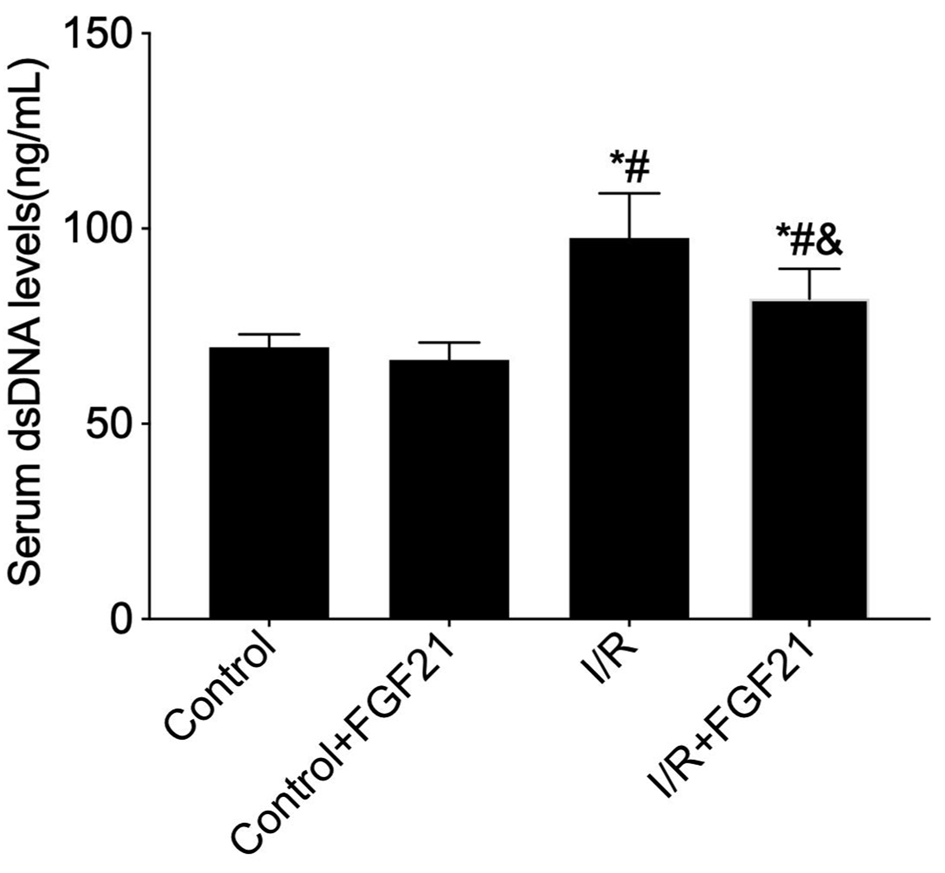 Click for large image | Figure 4. Measurement of peripheral serum dsDNA levels in FGF21 pretreated mice using the PicoGreen assay. *P < 0.05 versus control group; #P < 0.05 versus control + FGF21 group; &P < 0.05 versus I/R group. FGF21: fibroblast growth factor 21; I/R: ischemia/reperfusion. |
Inhibition of AMPK exacerbates myocardial tissue I/R injury
In order to investigate the mechanism by which FGF21 inhibited the expression of NETs induced by myocardial I/R injury, we first observed myocardial tissues after the inhibition of AMPK during I/R injury via H&E staining. In the I/R injury group, myocardial cells exhibited abnormal morphology, disorganization, and infiltration by a high quantity of inflammatory cells. Conversely, these pathological changes were significantly exacerbated in I/R-injured mice pretreated with compound C, an inhibitor of AMPK (Fig. 5). This study demonstrated that the inhibition of AMPK exacerbated myocardial I/R injury.
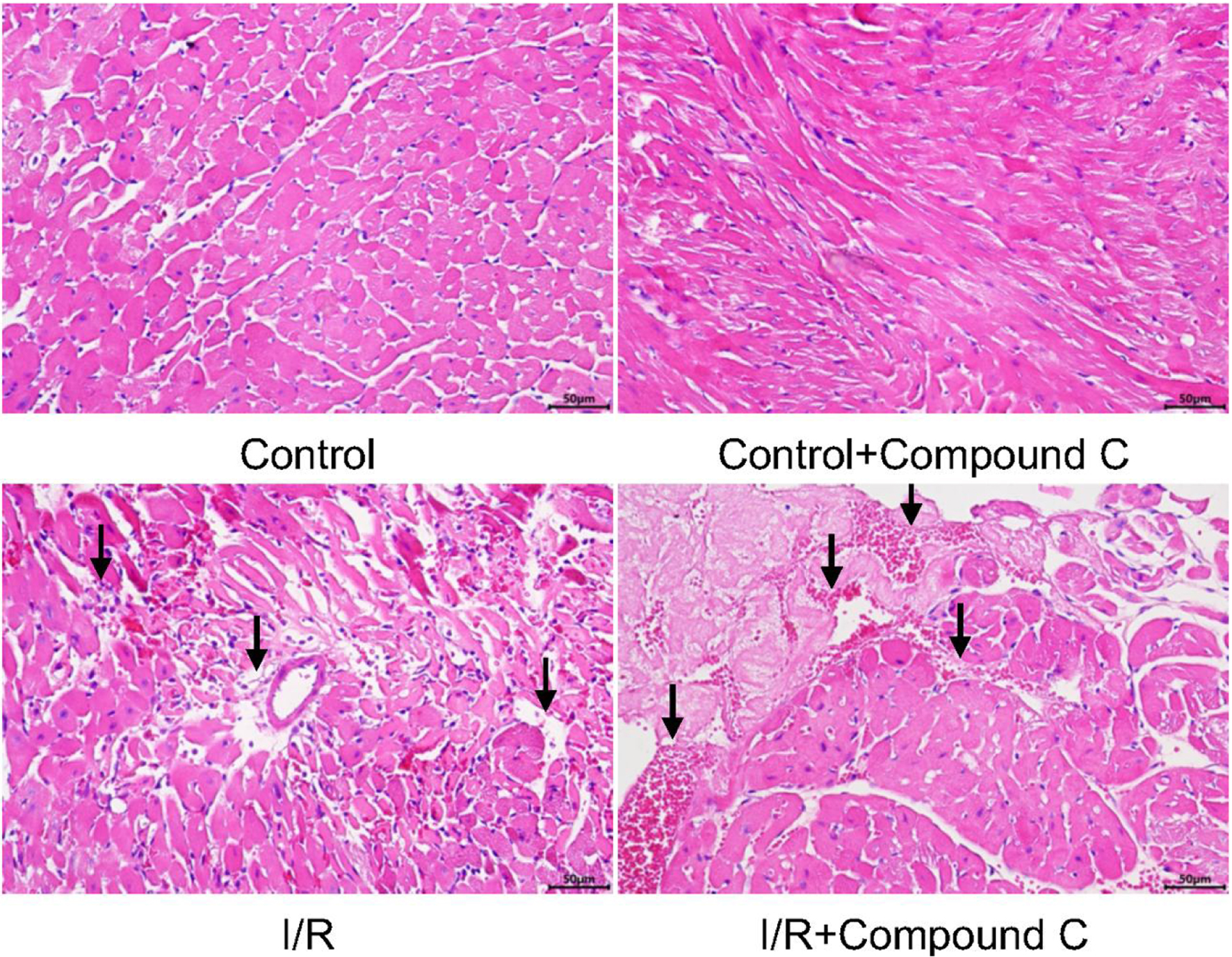 Click for large image | Figure 5. Myocardial tissue after myocardial I/R injury in mice pretreated with AMPK inhibitor (H&E staining, × 400). The black arrows represent obvious pathological changes in myocardial tissue. AMPK: AMP-activated protein kinase; H&E: hematoxylin and eosin; I/R: ischemia/reperfusion. |
Increased expression of NETs after AMPK inhibition
The expression of NETs in the myocardium of mice pretreated with an AMPK inhibitor was detected by immunofluorescence. Immunofluorescence revealed almost no histone H3 or MPO expression in the myocardial tissues of the control and control + compound C groups, indicating the absence of NET expression. In contrast, NET expression was greater in I/R-injured mice pretreated with compound C, compared to those in the I/R group (Fig. 6).
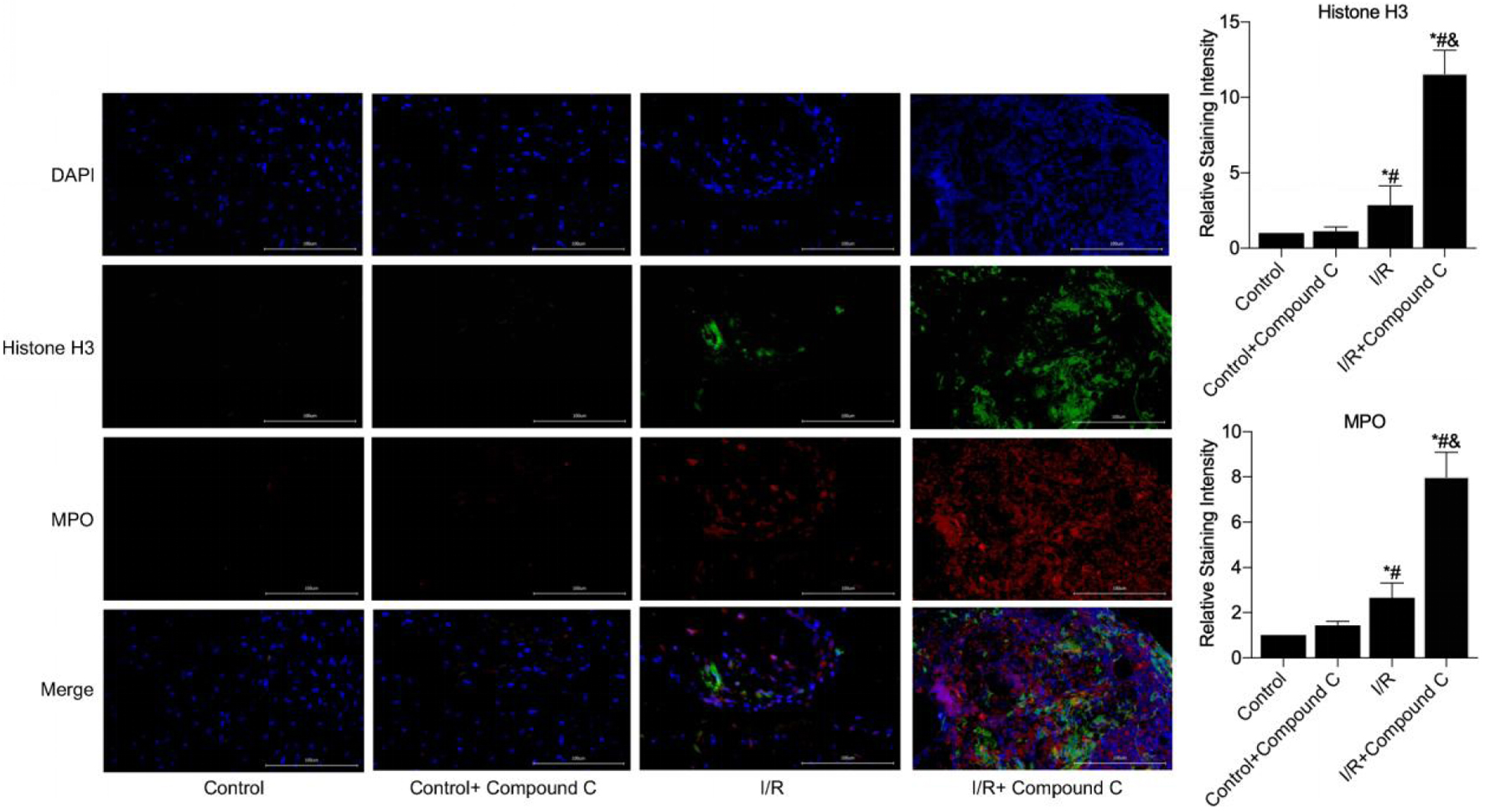 Click for large image | Figure 6. NETs formation in myocardial necrotic tissues was detected after the establishment of a myocardial I/R injury model in compound C pretreated mice (immunofluorescence, × 400). *P < 0.05 versus control group; #P < 0.05 versus control + FGF21 group; &P < 0.05 versus I/R group. FGF21: fibroblast growth factor 21; I/R: ischemia/reperfusion; NETs: neutrophil extracellular traps. |
Next, NET levels were quantified by PicoGreen and found to be significantly greater in the serum of the mice in the I/R group compared to those in the control group (103.03 ± 21.79 vs. 75.08 ± 8.03, P < 0.05). In contrast, NET levels were significantly greater in the I/R + compound C group compared to the I/R group (153.00 ± 20.11 vs. 103.03 ± 21.79, P < 0.05) (Fig. 7). However, there was no statistically significant difference in NET levels between the control and control + compound C groups. These results suggest that the inhibition of AMPK exacerbates NET expression during myocardial I/R injury.
 Click for large image | Figure 7. Measurement of peripheral serum dsDNA levels in compound C pretreated mice using the PicoGreen assay. *P < 0.05 versus control group; #P < 0.05 versus control + FGF21 group; &P < 0.05 versus I/R group. FGF21: fibroblast growth factor 21; I/R: ischemia/reperfusion. |
AMPK mediated the inhibition of myocardial I/R injury by FGF21
To further explore the relationship between FGF21 and AMPK, AMPK protein expression levels were measured by establishing an I/R injury model in mice preinjected with FGF21. AMPK protein expression levels were increased in the I/R group compared with those in the control group and further increased in the I/R + FGF21 group (P < 0.05) (Fig. 8a).
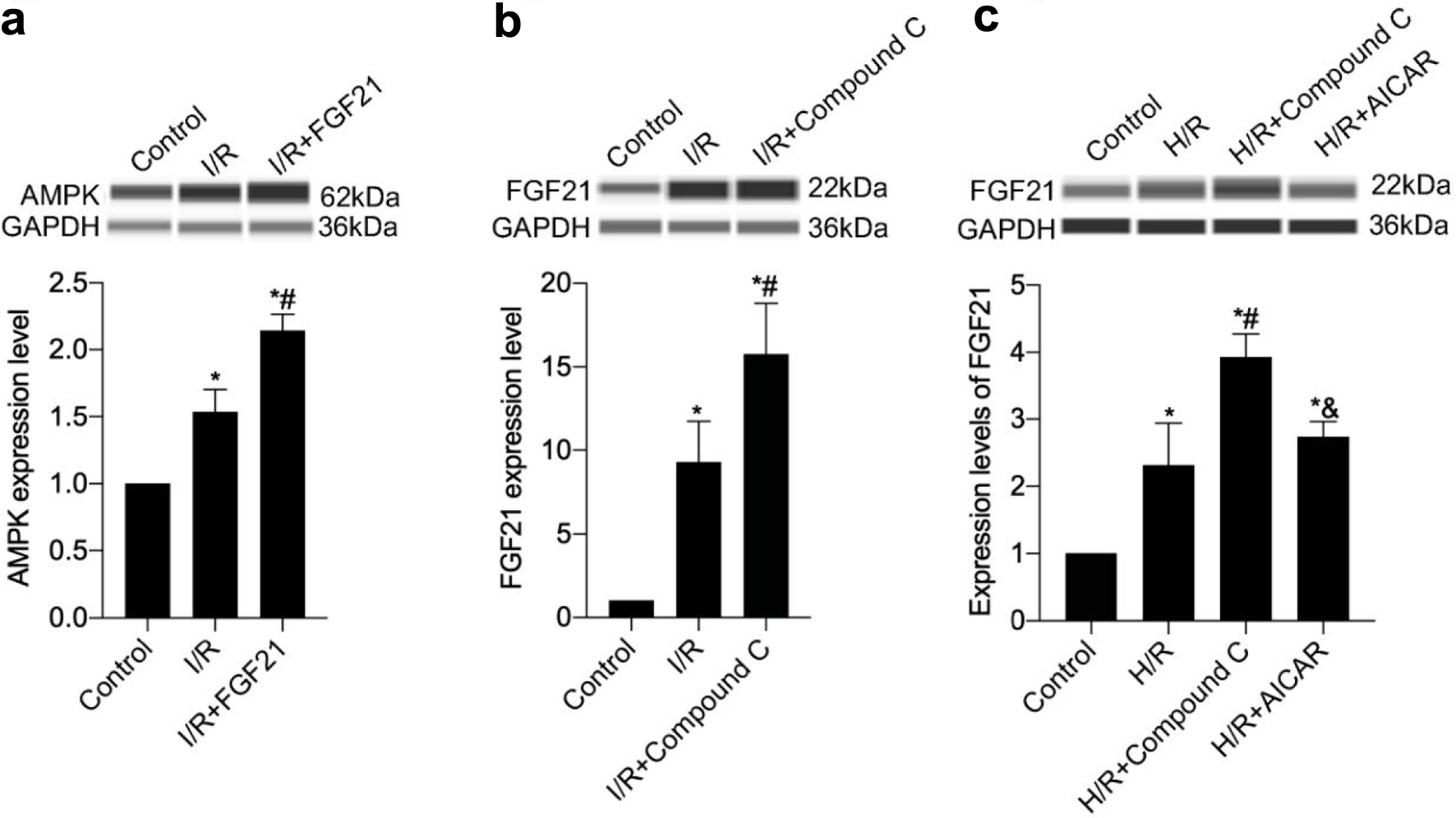 Click for large image | Figure 8. The interaction of FGF21 and AMPK was detected in a mouse model of myocardial I/R injury or an H/R model of H9c2 cells. (a) AMPK protein expression levels were measured in mice preinjected with FGF21. (b) FGF21 protein expression levels were measured in mice preinjected with AMPK inhibitor compound C. (c) FGF21 protein expression levels were measured in H9c2 cells pretreated with compound C, an inhibitor of AMPK, or with AICAR, an agonist. *P < 0.05 versus control; #P < 0.05 versus I/R group; &P < 0.05 versus H/R + compound C group. AMPK: AMP-activated protein kinase; FGF21: fibroblast growth factor 21; H/R: hypoxia/reoxygenation; I/R: ischemia/reperfusion. |
FGF21 protein expression levels were subsequently measured by establishing an I/R injury model in mice preinjected with compound C. FGF21 protein expression levels were significantly higher in the I/R group than in the control group and further increased in the I/R + compound C group (all P < 0.05) (Fig. 8b). These studies highlighted the involvement of AMPK in the cardioprotective effects of FGF21.
The H/R model was then established after pretreatment of H9c2 cells with compound C, an inhibitor of AMPK, and AICAR, an agonist of AMPK. Different hypoxia groups (2, 6, and 12 h) were set up with a reoxygenation time of 12 h to detect cellular activity. Compared with that of the control group, the cell activity of the H/R group decreased significantly (P < 0.05) (Fig. 9). Compared with that of the H/R group, the cell activity of the H/R + compound C group decreased further (P < 0.05), whereas the cell activity of the H/R + AICAR group increased significantly (P < 0.05). However, there was no statistically significant difference in cell activity among the control, control + compound C, and control + AICAR groups. Moreover, as the duration of hypoxia increased, the cell activity of each group gradually decreased and reached a minimum at 12 h of hypoxia. Consequently, 12 h of hypoxia was chosen as a critical time point to study further. This study demonstrated that AMPK was able to inhibit H/R-induced cardiomyocyte injury and had a cardioprotective effect.
Click for large image | Figure 9. AMPK inhibited H/R injury in H9c2 cells in a time-dependent manner. *P < 0.05 versus control; #P < 0.05 versus H/R group; &P < 0.05 versus H/R + compound C group. AMPK: AMP-activated protein kinase; H/R: hypoxia/reoxygenation. |
Finally, FGF21 protein expression levels were examined following pretreatment of H9c2 cells with the AMPK inhibitor compound C and the AMPK agonist AICAR. The FGF21 protein expression level in the H/R + compound C group was greater than that of the H/R group (P < 0.05), whereas the FGF21 protein expression level in the H/R + AICAR group was significantly lower than that of the H/R + compound C group (P < 0.05) (Fig. 8c). These findings suggest that AMPK plays an important role in the mechanism by which FGF21 inhibits myocardial H/R injury.
| Discussion | ▴Top |
The results of the present study suggested that FGF21 can attenuate myocardial I/R injury by inhibiting NET expression. Furthermore, the cardioprotective effect of FGF21 was mediated through the involvement of AMPK.
Numerous prior studies in this field have produced results that are consistent with our findings, demonstrating that FGF21 attenuates myocardial I/R injury, increases cardiomyocyte activity, and decreases the rate of cardiomyocyte apoptosis [19-21]. In a rat model of myocardial I/R injury, notable upregulation of NET expression was observed within the ischemic zone. Interestingly, deoxyribonuclease I was used to facilitate the degradation of NETs, which led to a reduction in myocardial infarct size and a significant improvement in left ventricular remodeling induced by I/R injury in rats [22]. In a mouse model of chronic pancreatitis, mice treated with FGF21 presented significantly reduced levels of MPO, a biomarker of NETs, and improved inflammatory states of the serum, pancreas, and peritoneal macrophages [23]. For the first time in this field of study, our current investigation made important discoveries within a myocardial I/R injury model, revealing that FGF21 can inhibit the expression of NETs induced by I/R injury, thereby attenuating myocardial I/R injury.
Previous studies have shown that FGF21 is an endocrine factor that can regulate energy metabolism and that AMPK, as an energy sensor, is a target for FGF21 signaling [24]. FGF21 activates AMPK both directly through the FGF receptor 1 (FGFR1)/Klotho-β complex and indirectly by inducing the expression of lipofuscin and corticosterone [25]. Once activated, AMPK plays a crucial role in regulating mitochondrial activity by stimulating sirtuin 1 (SIRT1) and peroxisome proliferator-activated receptor coactivator 1-α (PGC-1α) [26]. Furthermore, AMPK contributes to the maintenance of lipid homeostasis, protein synthesis regulation, and the promotion of glucose metabolism, all of which are aimed at sustaining optimal intracellular adenosine triphosphate levels [27].
In addition to its role in stimulating AMPK to regulate energy metabolism, FGF21 also has anti-inflammatory properties. Studies have shown that a lack of FGF21 increases susceptibility to inflammation, leading to local and systemic inflammatory responses, and supplementation with exogenous FGF21 has been shown to reverse elevated levels of these inflammatory markers [28-30]. In hypertensive and diabetic mouse models, the absence of FGF21 resulted in a pronounced exacerbation of the inflammatory response and oxidative stress. Conversely, the administration of FGF21 significantly alleviated these conditions by promoting the phosphorylation of AMPK, highlighting the crucial role of FGF21 in mitigating inflammatory and oxidative stress pathways [31, 32]. Subsequent investigations revealed that FGF21 exerts its anti-inflammatory, antioxidant, and inhibitory effects on myocardial remodeling and apoptosis through two distinct pathways. The first pathway involves the AMPK-AKT2-Nrf2 antioxidant pathway, whereas the second pathway operates through the AMPK-ACC-CPT-1 lipid-lowering pathway, both of which are mediated by AMPK [33].
Studies have shown that imbalances in the mechanisms that produce or eliminate NETs can lead to tissue damage, which is an important mechanism in autoimmune and inflammatory diseases that are closely linked to cardiovascular disease, and that inhibiting NET expression has been shown to be beneficial [34, 35]. Several studies have consistently demonstrated that AMPK, known for its anti-inflammatory effects, plays a pivotal role in the expression of NETs [36-39]. The activation of AMPK both inhibits NET expression and promotes NET clearance to reduce NET levels [40, 41]. However, inhibition of AMPK significantly increased the levels of NETs in the peripheral circulation and ischemic tissues [42].
It was found that statin and metformin, which are routinely applied in clinical practice, can activate AMPK, protect cardiac tissues from I/R-induced excessive damage, and improve clinical prognosis [43, 44]. In addition, clinical studies found that plasma FGF21 levels were significantly increased in AMI patients with spironolactone application, and further studies found that spironolactone could improve cardiac function and adverse cardiac remodeling by promoting the expression of FGF21 [45]. In summary, a variety of drugs that promote the expression of FGF21 and AMPK have the potential to be used as adjunctive therapeutic agents to existing reperfusion therapy.
The present study has several limitations. Owing to the limitations of funds and experimental equipment, cardiac ultrasonography was not performed in I/R-injured mice in this study, so it was not possible to judge the improvement in cardiac function in mice after the application of FGF21. Although this study suggests a possible mechanism by which FGF21 inhibits the expression of NETs through the activation of AMPK, the actual biological mechanism is likely to be more complex, and other unconsidered signaling pathways or cell types may also be involved.
Conclusions
In the present study, we observed a significant elevation in NET levels in both serum and myocardial tissue during myocardial I/R injury. Notably, the administration of FGF21 significantly reversed the increase in NET levels. Subsequent studies delved into the underlying mechanisms, suggesting that FGF21 may inhibit I/R injury-induced NET expression through the activation of AMPK. These findings highlight a potential therapeutic role for FGF21 in mitigating NET-mediated responses in the context of myocardial I/R injury.
Acknowledgments
None to declare.
Financial Disclosure
This research was supported by the development fund of Affiliated Hospital of Xuzhou Medical University (grant no. XYFY2021029), Medical Research Program of Jiangsu Provincial Health and Wellness Commission (grant no. Z2021042), and Jiangyin Young and Middle-aged Reserve Excellent Talents Program (grant no. JYROYT202309).
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
Not applicable.
Author Contributions
LYG, CGJ, ZZS, and JYC conducted the methodology and wrote the manuscript. WLJ and ZWX performed the analyses and reviewed the manuscript. WZL and HZ designed the experiment and coordinated.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
AMI: acute myocardial infarction; AMP: adenosine monophosphate; AMPK: AMP-activated protein kinase; FGF21: fibroblast growth factor 21; H/R: hypoxia/reoxygenation; I/R: ischemia/reperfusion; MACE: major adverse cardiac event; MPO: myeloperoxidase; NETs: neutrophil extracellular traps; PCI: percutaneous coronary intervention; SPF: specific-pathogen-free
| References | ▴Top |
- Bhatt DL, Lopes RD, Harrington RA. Diagnosis and treatment of acute coronary syndromes: a review. JAMA. 2022;327(7):662-675.
doi pubmed - Ibanez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65(14):1454-1471.
doi pubmed - Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth. 2012;16(3):123-132.
doi pubmed pmc - Frohlich GM, Meier P, White SK, Yellon DM, Hausenloy DJ. Myocardial reperfusion injury: looking beyond primary PCI. Eur Heart J. 2013;34(23):1714-1722.
doi pubmed - Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123(1):92-100.
doi pubmed pmc - Barzyc A, Lysik W, Slyk J, Kuszewski M, Zarebinski M, Wojciechowska M, Cudnoch-Jedrzejewska A. Reperfusion injury as a target for diminishing infarct size. Med Hypotheses. 2020;137:109558.
doi pubmed - Puhl SL, Steffens S. Neutrophils in Post-myocardial Infarction Inflammation: Damage vs. Resolution? Front Cardiovasc Med. 2019;6:25.
doi pubmed pmc - Hoppenbrouwers T, Autar ASA, Sultan AR, Abraham TE, van Cappellen WA, Houtsmuller AB, van Wamel WJB, et al. In vitro induction of NETosis: comprehensive live imaging comparison and systematic review. PLoS One. 2017;12(5):e0176472.
doi pubmed pmc - Fousert E, Toes R, Desai J. Neutrophil Extracellular Traps (NETs) Take the Central Stage in Driving Autoimmune Responses. Cells. 2020;9(4):915.
doi pubmed pmc - Mangold A, Alias S, Scherz T, Hofbauer M, Jakowitsch J, Panzenbock A, Simon D, et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res. 2015;116(7):1182-1192.
doi pubmed - Riegger J, Byrne RA, Joner M, Chandraratne S, Gershlick AH, Ten Berg JM, Adriaenssens T, et al. Histopathological evaluation of thrombus in patients presenting with stent thrombosis. A multicenter European study: a report of the prevention of late stent thrombosis by an interdisciplinary global European effort consortium. Eur Heart J. 2016;37(19):1538-1549.
doi pubmed pmc - Langseth MS, Opstad TB, Bratseth V, Solheim S, Arnesen H, Pettersen AA, Seljeflot I, et al. Markers of neutrophil extracellular traps are associated with adverse clinical outcome in stable coronary artery disease. Eur J Prev Cardiol. 2018;25(7):762-769.
doi pubmed - Novotny J, Oberdieck P, Titova A, Pelisek J, Chandraratne S, Nicol P, Hapfelmeier A, et al. Thrombus NET content is associated with clinical outcome in stroke and myocardial infarction. Neurology. 2020;94(22):e2346-e2360.
doi pubmed - Hally KE, Parker OM, Brunton-O'Sullivan MM, Harding SA, Larsen PD. Linking neutrophil extracellular traps and platelet activation: a composite biomarker score for predicting outcomes after acute myocardial infarction. Thromb Haemost. 2021;121(12):1637-1649.
doi pubmed - Doring Y, Soehnlein O, Weber C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ Res. 2017;120(4):736-743.
doi pubmed - Li YW, Chen SX, Yang Y, Zhang ZH, Zhou WB, Huang YN, Huang ZQ, et al. Colchicine inhibits nets and alleviates cardiac remodeling after acute myocardial infarction. Cardiovasc Drugs Ther. 2024;38(1):31-41.
doi pubmed - Gu L, Jiang W, Jiang W, Xu Z, Li W, Zhang H. Elevated serum FGF21 levels predict heart failure during hospitalization of STEMI patients after emergency percutaneous coronary intervention. PeerJ. 2023;11:e14855.
doi pubmed pmc - Gu L, Jiang W, Qian H, Zheng R, Li W. Elevated serum FGF21 predicts the major adverse cardiovascular events in STEMI patients after emergency percutaneous coronary intervention. PeerJ. 2021;9:e12235.
doi pubmed pmc - Hu S, Cao S, Liu J. Role of angiopoietin-2 in the cardioprotective effect of fibroblast growth factor 21 on ischemia/reperfusion-induced injury in H9c2 cardiomyocytes. Exp Ther Med. 2017;14(1):771-779.
doi pubmed pmc - Hu S, Cao S, Tong Z, Liu J. FGF21 protects myocardial ischemia-reperfusion injury through reduction of miR-145-mediated autophagy. Am J Transl Res. 2018;10(11):3677-3688.
pubmed pmc - Cong WT, Ling J, Tian HS, Ling R, Wang Y, Huang BB, Zhao T, et al. Proteomic study on the protective mechanism of fibroblast growth factor 21 to ischemia-reperfusion injury. Can J Physiol Pharmacol. 2013;91(11):973-984.
doi pubmed - Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang YD, Ma YQ, et al. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol. 2015;308(5):H500-509.
doi pubmed - Wang N, Zhao TT, Li SM, Li YH, Wang YJ, Li DS, Wang WF. Fibroblast growth factor 21 ameliorates pancreatic fibrogenesis via regulating polarization of macrophages. Exp Cell Res. 2019;382(1):111457.
doi pubmed - Salminen A, Kaarniranta K, Kauppinen A. Regulation of longevity by FGF21: Interaction between energy metabolism and stress responses. Ageing Res Rev. 2017;37:79-93.
doi pubmed - Zhang N, Liu C, Zhang Y, Xu D, Gui L, Lu Y, Zhang Q. Liraglutide regulates lipid metabolism via FGF21- LKB1- AMPK- ACC1 pathway in white adipose tissues and macrophage of type 2 diabetic mice. Biochem Biophys Res Commun. 2021;548:120-126.
doi pubmed - Chau MD, Gao J, Yang Q, Wu Z, Gromada J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc Natl Acad Sci U S A. 2010;107(28):12553-12558.
doi pubmed pmc - Wu S, Zou MH. AMPK, Mitochondrial function, and cardiovascular disease. Int J Mol Sci. 2020;21(14):4987.
doi pubmed pmc - Lin Z, Pan X, Wu F, Ye D, Zhang Y, Wang Y, Jin L, et al. Fibroblast growth factor 21 prevents atherosclerosis by suppression of hepatic sterol regulatory element-binding protein-2 and induction of adiponectin in mice. Circulation. 2015;131(21):1861-1871.
doi pubmed pmc - Sharma S, Dixon T, Jung S, Graff EC, Forney LA, Gettys TW, Wanders D. Dietary Methionine Restriction Reduces Inflammation Independent of FGF21 Action. Obesity (Silver Spring). 2019;27(8):1305-1313.
doi pubmed pmc - Song JJ, Yang M, Liu Y, Song JW, Liu XY, Miao R, Zhang ZZ, et al. Elabela prevents angiotensin II-induced apoptosis and inflammation in rat aortic adventitial fibroblasts via the activation of FGF21-ACE2 signaling. J Mol Histol. 2021;52(5):905-918.
doi pubmed pmc - Kang K, Xu P, Wang M, Chunyu J, Sun X, Ren G, Xiao W, et al. FGF21 attenuates neurodegeneration through modulating neuroinflammation and oxidant-stress. Biomed Pharmacother. 2020;129:110439.
doi pubmed - Weng HC, Lu XY, Xu YP, Wang YH, Wang D, Feng YL, Chi Z, et al. Fibroblast growth factor 21 attenuates salt-sensitive hypertension-induced nephropathy through anti-inflammation and anti-oxidation mechanism. Mol Med. 2021;27(1):147.
doi pubmed pmc - Yang H, Feng A, Lin S, Yu L, Lin X, Yan X, Lu X, et al. Fibroblast growth factor-21 prevents diabetic cardiomyopathy via AMPK-mediated antioxidation and lipid-lowering effects in the heart. Cell Death Dis. 2018;9(2):227.
doi pubmed pmc - Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol. 2012;189(6):2689-2695.
doi pubmed pmc - Li Y, Liu J, Cui Y, Cao Y, Xu P, Kan X, Guo W, et al. Sodium butyrate attenuates bovine mammary epithelial cell injury by inhibiting the formation of neutrophil extracellular traps. Int Immunopharmacol. 2022;110:109009.
doi pubmed - Song Y, Zhang Y, Zhang P, Yu P, Shang X, Lu Y, Li Y, et al. Effects of fluorine on neutrophil extracellular trap formation through regulating AMPK/p38 signaling pathway. Oxid Med Cell Longev. 2021;2021:6693921.
doi pubmed pmc - Ma Y, Li S, Ye S, Tang S, Hu D, Wei L, Xiao F. Hexavalent chromium inhibits the formation of neutrophil extracellular traps and promotes the apoptosis of neutrophils via AMPK signaling pathway. Ecotoxicol Environ Saf. 2021;223:112614.
doi pubmed - Ong CW, Elkington PT, Brilha S, Ugarte-Gil C, Tome-Esteban MT, Tezera LB, Pabisiak PJ, et al. Neutrophil-derived MMP-8 drives AMPK-dependent matrix destruction in human pulmonary tuberculosis. PLoS Pathog. 2015;11(5):e1004917.
doi pubmed pmc - Rodriguez C, Munoz M, Contreras C, Prieto D. AMPK, metabolism, and vascular function. FEBS J. 2021;288(12):3746-3771.
doi pubmed - Gregoire M, Uhel F, Lesouhaitier M, Gacouin A, Guirriec M, Mourcin F, Dumontet E, et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur Respir J. 2018;52(2):1702590.
doi pubmed - Chiang N, Sakuma M, Rodriguez AR, Spur BW, Irimia D, Serhan CN. Resolvin T-series reduce neutrophil extracellular traps. Blood. 2022;139(8):1222-1233.
doi pubmed pmc - Wu ML, Zou X, Chen XY, Ma KT, Chen C, Yu NW, Yu L, et al. Phthalide derivative CD21 regulates the platelet- neutrophil extracellular trap-thrombin axis and protects against ischemic brain injury in rodents. Int Immunopharmacol. 2023;114:109547.
doi pubmed - Hua P, Liu J, Tao J, Zou R, Lin X, Zhang D, Yang S. Efficacy and mechanism of preoperative simvastatin therapy on myocardial protection after extracorporeal circulation. Biomed Res Int. 2017;2017:6082430.
doi pubmed pmc - Higgins L, Palee S, Chattipakorn SC, Chattipakorn N. Effects of metformin on the heart with ischaemia-reperfusion injury: Evidence of its benefits from in vitro, in vivo and clinical reports. Eur J Pharmacol. 2019;858:172489.
doi pubmed - Sun JY, Du LJ, Shi XR, Zhang YY, Liu Y, Wang YL, Chen BY, et al. An IL-6/STAT3/MR/FGF21 axis mediates heart-liver cross-talk after myocardial infarction. Sci Adv. 2023;9(14):eade4110.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.