

| Cardiology Research, ISSN 1923-2829 print, 1923-2837 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cardiol Res and Elmer Press Inc |
| Journal website http://www.cardiologyres.org |
Case Report
Volume 9, Number 5, October 2018, pages 324-329
Holt-Oram Syndrome With Multiple Cardiac Abnormalities
Marilena Renata Spiridona, Antoniu Octavian Petrisa, b, Eusebiu Vlad Gorduzac, Anca Sabina Petrasa, d, Roxana Popescuc, Lavinia Cabac
aCardiology Department, “St. Spiridon” Emergency Hospital, Iasi, Romania
bCardiology Department, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania
cMedical Genetics Department, “Grigore T. Popa” University of Medicine and Pharmacy, Iasi, Romania
dCorresponding Author: Anca Sabina Petras, Cardiology Department, “St. Spiridon” Emergency Hospital, Iasi, Romania
Manuscript submitted July 30, 2018, accepted September 5, 2018
Short title: Holt-Oram Syndrome
doi: https://doi.org/10.14740/cr767w
| Abstract | ▴Top |
Holt-Oram syndrome (HOS) is a rare monogenic disorder characterized by upper limb abnormalities, congenital heart defects and/or conduction abnormalities. It is determined by mutations of TBX5 gene and is inherited in an autosomal dominant manner. Penetrance is complete, but variable expressivity is present, which gives sometimes diagnostic difficulties. Our case is a young adult with a personal history of preaxial polydactyly operated in infancy, multiple cardiac malformations (atrial septal defect, bicuspid aortic valve, left ventricular non-compaction) and radiologic findings consistent with HOS. Family history is negative for HOS. In conclusion, we present a case of HOS diagnosed in the adult period to highlight the diagnostic problems for the proband and the family and the importance of an early diagnostic.
Keywords: Congenital heart defects; Preaxial polydactyly; Carpal bones; Atrial septal defect; Holt-Oram syndrome
| Introduction | ▴Top |
Holt-Oram syndrome (HOS; MIM 142900) is a rare monogenic disorder characterized by upper limb abnormalities, congenital heart defects (CHD) and/or conduction abnormalities [1]. HOS is the most common heart-hand syndrome (defined by congenital heart defects associated with skeletal abnormalities of the upper limb) usually associated with an atrial septal defect (ASD). Uncommon subtypes of heart-hand syndrome are Tabatznik syndrome (type II; arrhythmias and brachytelephalangy) and Spanish variant (type III; arrhythmia and brachydactyly type C) and potential Slovenian type (type IV; arrhythmia, dilated cardiomyopathy, brachydactyly) [2].
HOS was first described by Mary Clayton Holt and Samuel Oram in 1960, when they reported members of a family with thumb abnormalities and ASD. The birth prevalence of HOS is estimated to be about 0.7 in 100,000 births without gender differences [3].
HOS is an autosomal dominant condition, being caused by a mutation in TBX5 gene (located on chromosome 12q24.1). Majority of the mutations are de novo. TBX5 is expressed especially in the heart and forelimb, but also in lungs and eyes [4]. TBX5 is a protein coding gene that consists in nine coding exons. The gene belongs to T-box domain. These T-box domains contain about 180 amino acid residues. These genes encode transcription factors involved in development regulation. Most mutations described in HOS are mutations within the T-box domain. The gene expression of TBX5 is different across the heart tube being maximal at the inflow tract of the heart [5]. The other cardiac regions with TBX5 expression are: sinus venosus, atria, atrioventricular canal, left ventricle (LV), and right ventricle trabeculae. For this reason, there are effects on cardiac conduction system in atrioventricular node, atria, His bundle and bundle branches [5, 6]. Transcription factor has not effects on selected clinical outcomes (survival, growth, neurodevelopment, ventricular function) [7].
In heart development, many gene networks are involved and TBX5 interacts with some of them. The product of TBX5 gene has a combined action with GATA4 gene and presents a synergistic action with Nkx2-5 gene in coactivation of ANF gene by binding to their promoter region (cardiac-specific natriuretic precursor peptide type A) [8]. Another mechanism for TBX5 in heart development is the interaction TBX5-nucleosome remodeling and deacetylase repressor complex with consequences in cardiac septation [9].
TBX5 gene is involved in forelimb specification and outgrowth and is expressed from the earliest stages of forelimb development. TBX5 plays also a role in dorsoventral patterning of the eye, but pathogenic variants are not known to result in ocular abnormalities in humans [10]. Recent studies suggest that the KLF13 gene could be a modifying gene for TBX5 gene. The Tbx5 heterozygote mice with Klf13 heterozygote status shows no detectable cardiac defects, but loss of a second Klf13 allele determines at the same mice the appearance of cardiac defects specific for HOS [11].
Mutations in TBX5 gene were identified in more than 70% of cases with clinical signs of HOS. More than 70 mutations have been described in HOS. Pathogenic variants described were: missense or nonsense substitutions, large deletion of multiple exons, frameshift mutation, intragenic duplications [12]. The consequences of the mutations are haploinsufficiency, transcripts with a low DNA binding activity, or dominant-negative effect on downstream targets [12].
The disease is a pleiotropic disorder with complete penetrance, but with a variable expressivity, even in the same family.
The diagnosis of HOS is essentially clinical. Clinical diagnostic criteria of HOS include CHD and/or conduction defects and preaxial radial anomalies in at least one upper limb identified by X-ray examination. The cardiac and upper limb abnormalities may vary in presentation, from structural cardiac anomalies to conduction disorders, respectively from an abnormal carpal bone or triphalangeal thumb to bilateral phocomelia. To note that the upper limb abnormalities can be very subtle, sometimes the only abnormality is a delayed carpal bone age on wrist radiographs [13].
The main clinical characteristics are: upper-limb malformation, CHD, cardiac conduction disease and for each category there are many variants of presentation (Table 1 [1, 13]). All patients present a forelimb anomaly and 75% of patients with HOS have a structural cardiac anomaly, isolated septal defects, usually ASD or ventricular septal defects (VSD) involving the trabecular septum, being the most common. Approximately 25% of patients have complex CHD [1].
 Click to view | Table 1. Clinical Characteristics in HOS [1, 13] |
The purpose of this paper is to highlight the importance of an early diagnostic, to prevent the complications and to identify other risk family members.
| Case Report | ▴Top |
A 18 years and 5 months male presented with pediatric history of polydactyly of the right hand that was operated at the age of 4 years (surgical scar of the thumb) (Fig.1), dilated cardiomyopathy and severe mitral regurgitation discovered at the age of 2 months; after the investigation of a systolic murmur, later complicated with a left ventricular (LV) thrombus, who was then diagnosed with valvular changes (thickened aortic valve, mitral valve (MV) prolapse) and ostium secundum ASD with left to right shunt at the age of 3 years.
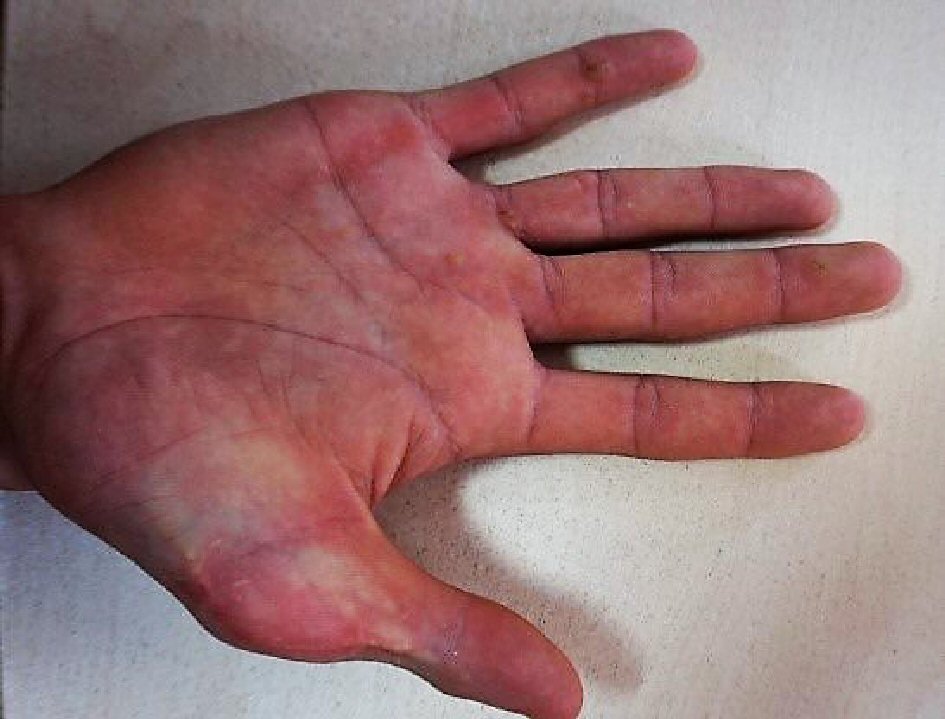 Click for large image | Figure 1. Right hand: surgical scar of the thumb. |
When addressing to the cardiology clinic, the patient had no symptoms and normal effort tolerance, declaratory. Clinically, he had perioral cyanosis, blood pressure of 110/70 mm Hg, heart rate (HR) of 85 bpm and a systolic 2/6 murmur at the apex.
The electrocardiogram (ECG) reveals a HR of 85 bpm, a heart axis of +60°, peaked P waves (P pulmonale) in II, III, aVF and LV hypertrophy (Sokolow Lyon criteria) (Fig. 2).
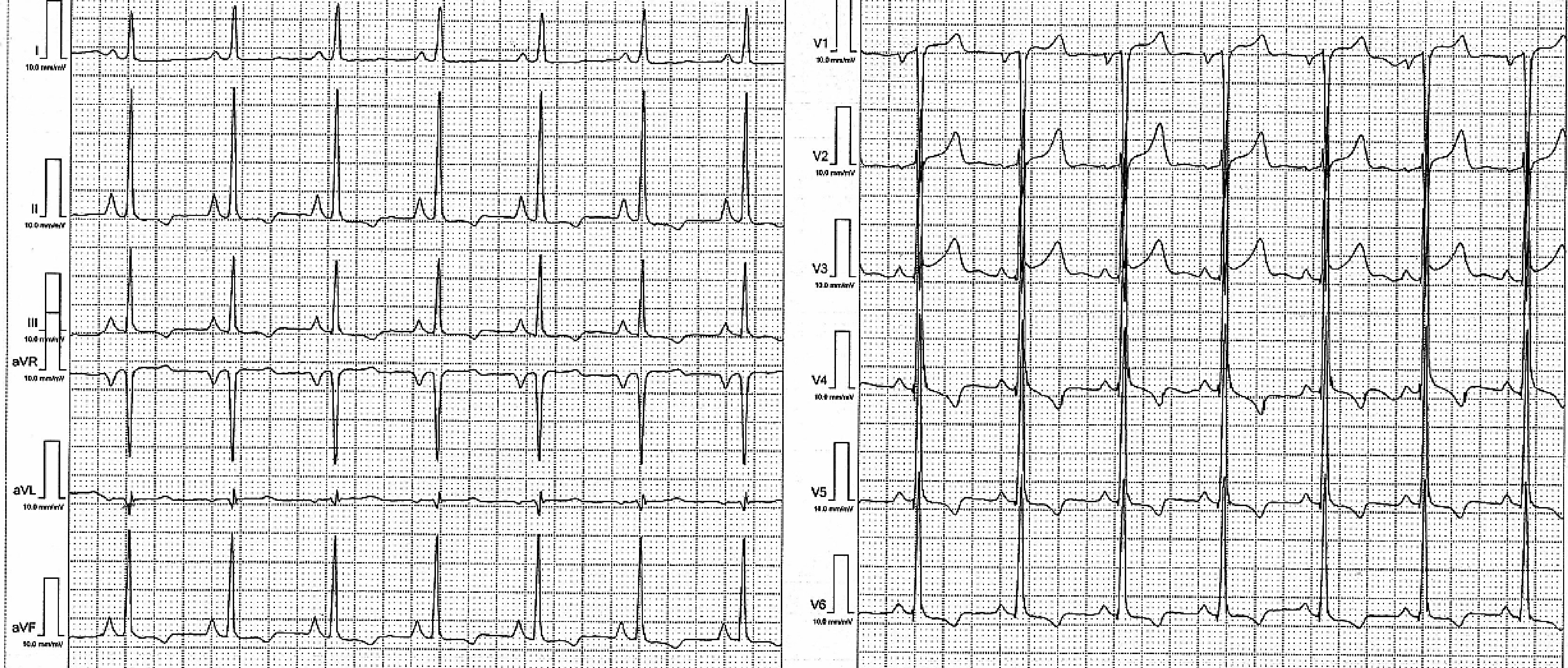 Click for large image | Figure 2. 12-lead ECG: sinus rhythm of 85 bpm, AQRS +60°, LV hypertrophy and P pulmonale. |
The transthoracic echocardiogram (TTE) shows a mildly dilated LV (adjusted for body surface area, 3.2 cm/m2), but with the rest of cardiac chambers within normal limits, a thickened MV, mainly the anterior MV, with an adjacent calcified cord. The MV closes with grade 2 - 3 mitral regurgitation with eccentric jet pointing to the posterior wall of the left atrium, but without the appearance of MV prolapse. The aortic valve appears tricuspid, thickened. An ostium secundum ASD is noted, with left to right shunt (Fig. 3). The TTE also reveals trabeculations of the LV lateral wall (Fig. 4), septal and anterior wall hypokinesis, and global LV ejection fraction of 50%.
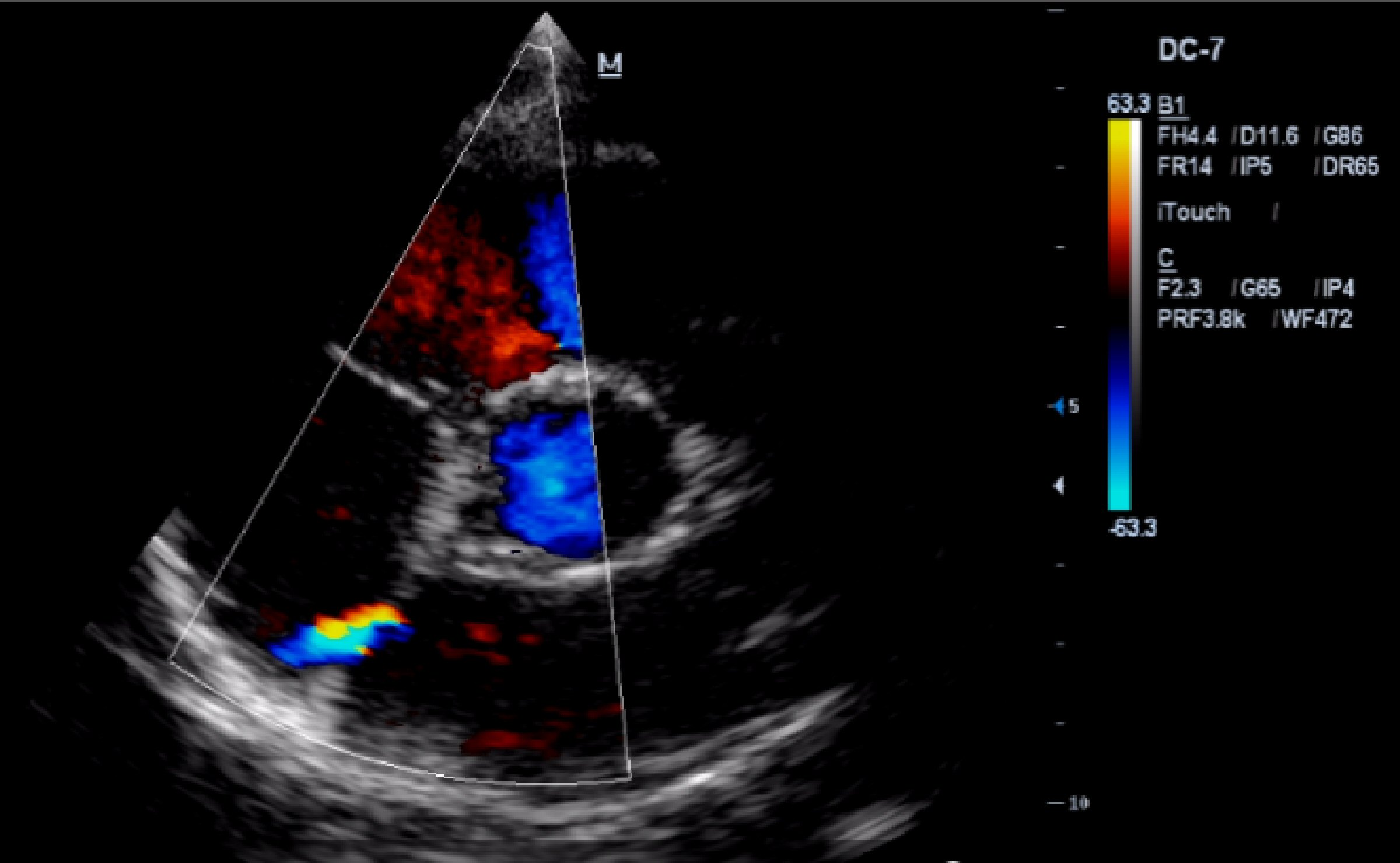 Click for large image | Figure 3. TTE (parasternal short axis view): ASD with left to right shunt. |
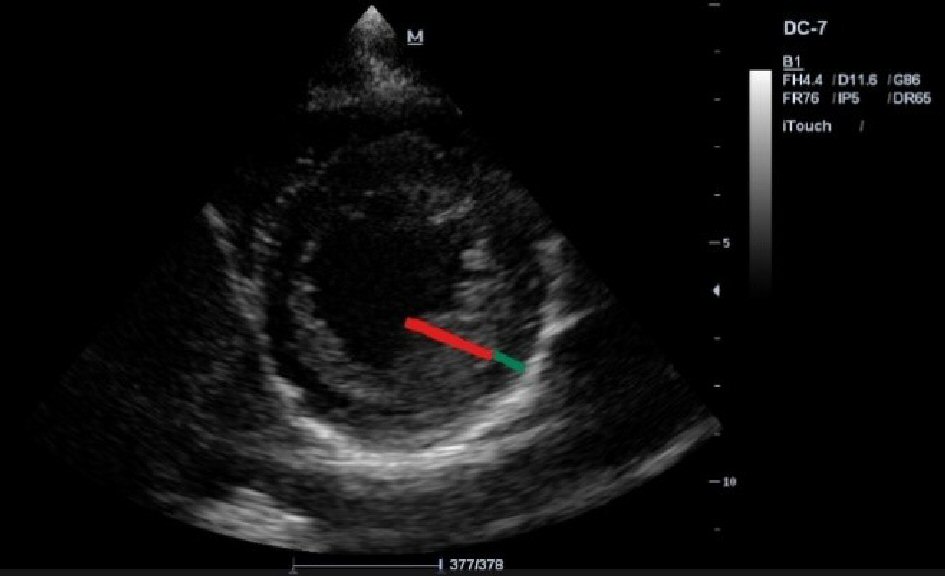 Click for large image | Figure 4. TTE (parasternal short axis view): trabeculations of the LV lateral wall with a non-compacted endocardial layer and a thin compacted layer. |
Due to the multiple changes that have been found on TTE, we turned to transesophageal echocardiography (TEE) for a better assessment of associated lesions and to visualize a possible MV prolapse as etiology of the mitral regurgitation. Thus, it has been noticed that the aortic valve was actually bicuspid, the right coronary cusp and the noncoronary cusp being welded together by a hyperechoic raphe (Fig. 5). No MV prolapse aspect was noticed. For the diagnosis of ASD, a bubble contrast echocardiogram was performed with the detection of left to right shunt by negative contrast effect (Fig. 6).
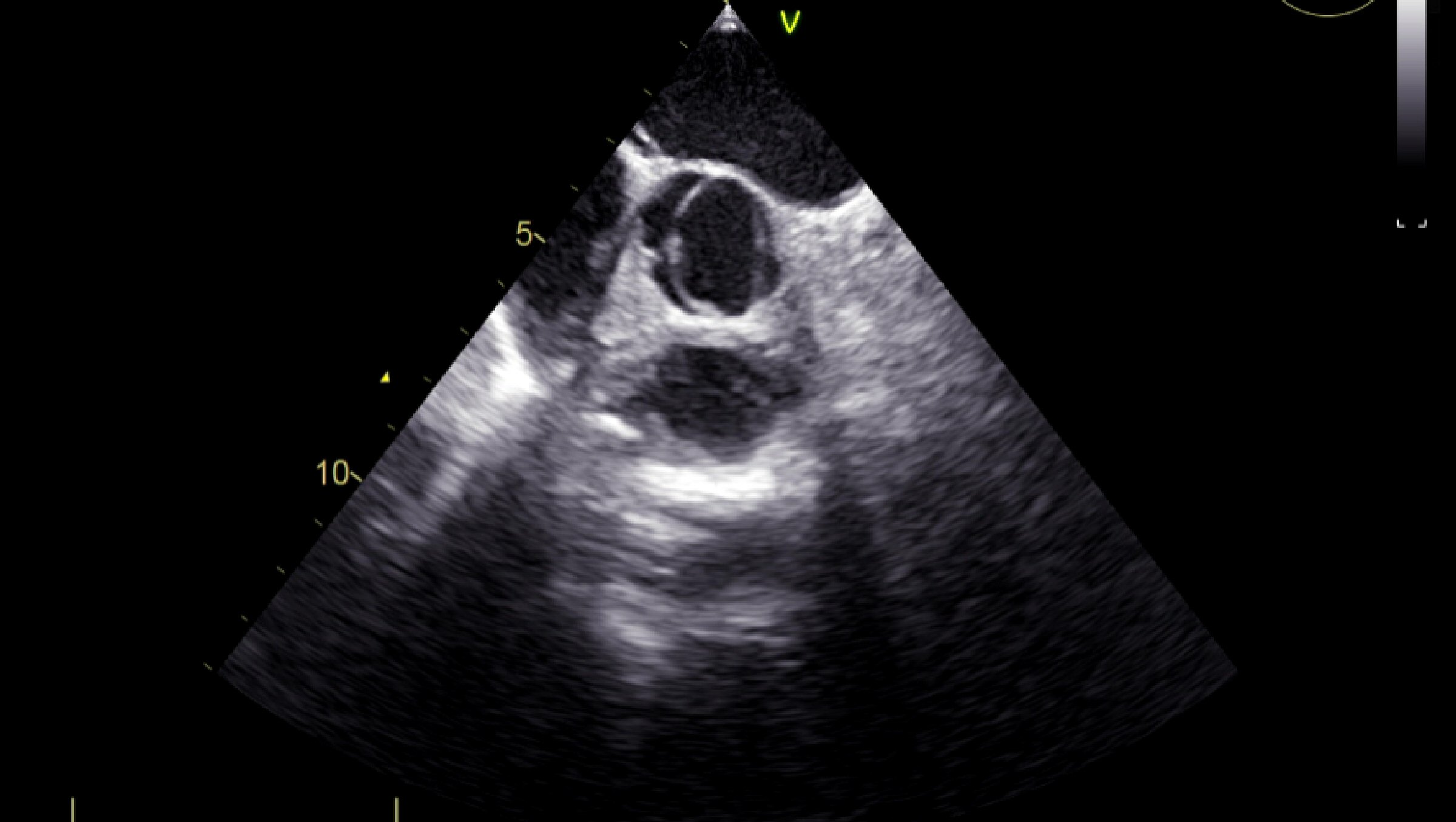 Click for large image | Figure 5. TEE (short axis view): bicuspid aortic valve. |
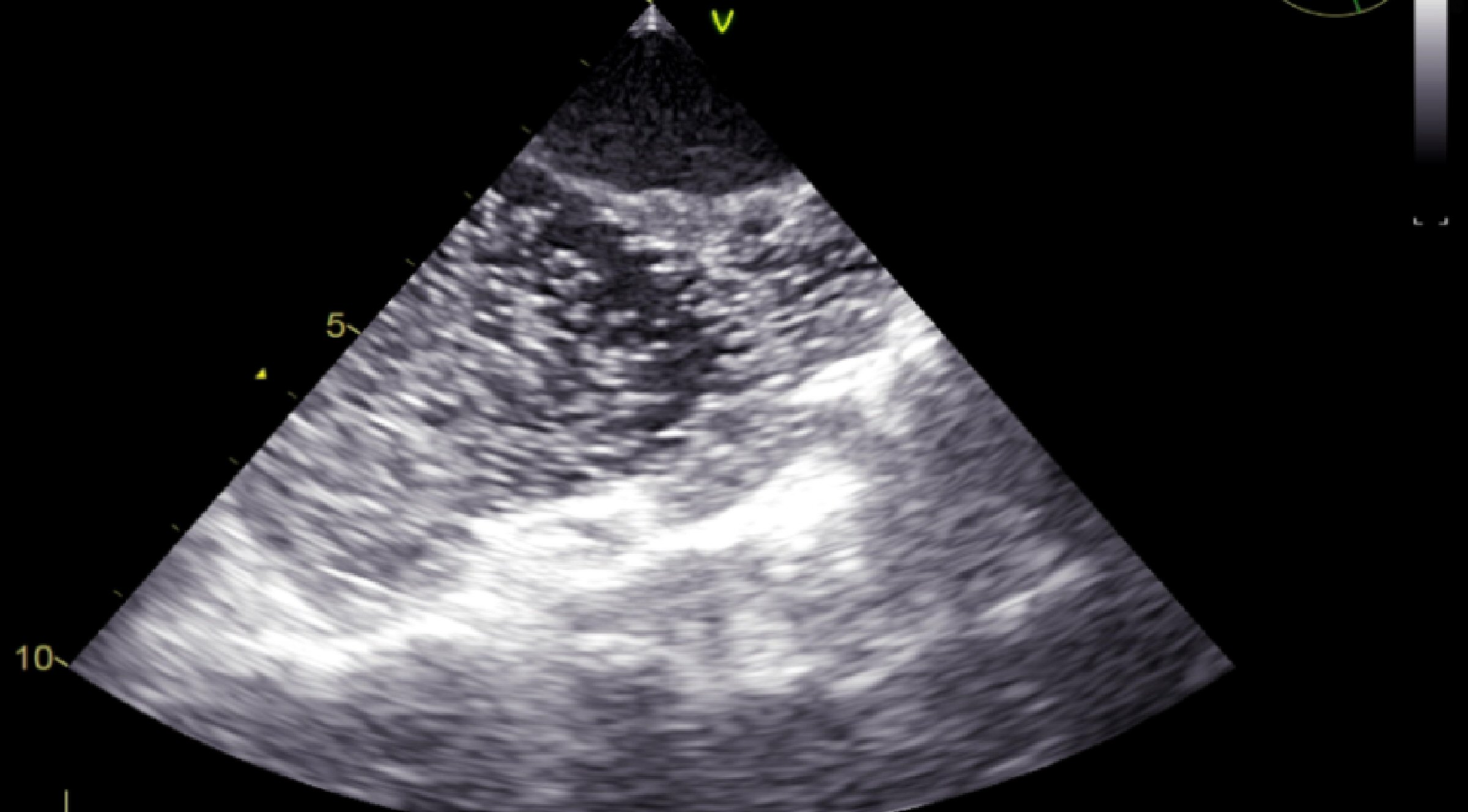 Click for large image | Figure 6. TEE (bicaval view): negative contrast effect showing the left to right shunt in ASD. |
Considering the multiple cardiac abnormalities found in this 18-year patient, we took a closer look at the antecedents of polydactyly of the hand, thinking of a genetic disorder that affects both heart and hand. We continued with radiologic investigation of upper limbs and genetic testing by multiplex ligation-dependent probe amplification (MLPA) for TBX5 gene, in this purpose. The radiographic imaging of the hands showed a delayed bone age, corresponding to a 16-year patient, small sized bilateral trapezium and trapezoid bones that are partly overlapped, hypertrophic bilateral pisiform bones and a metacarpophalangeal angle over 5° at the third and forth digits (Fig. 7).
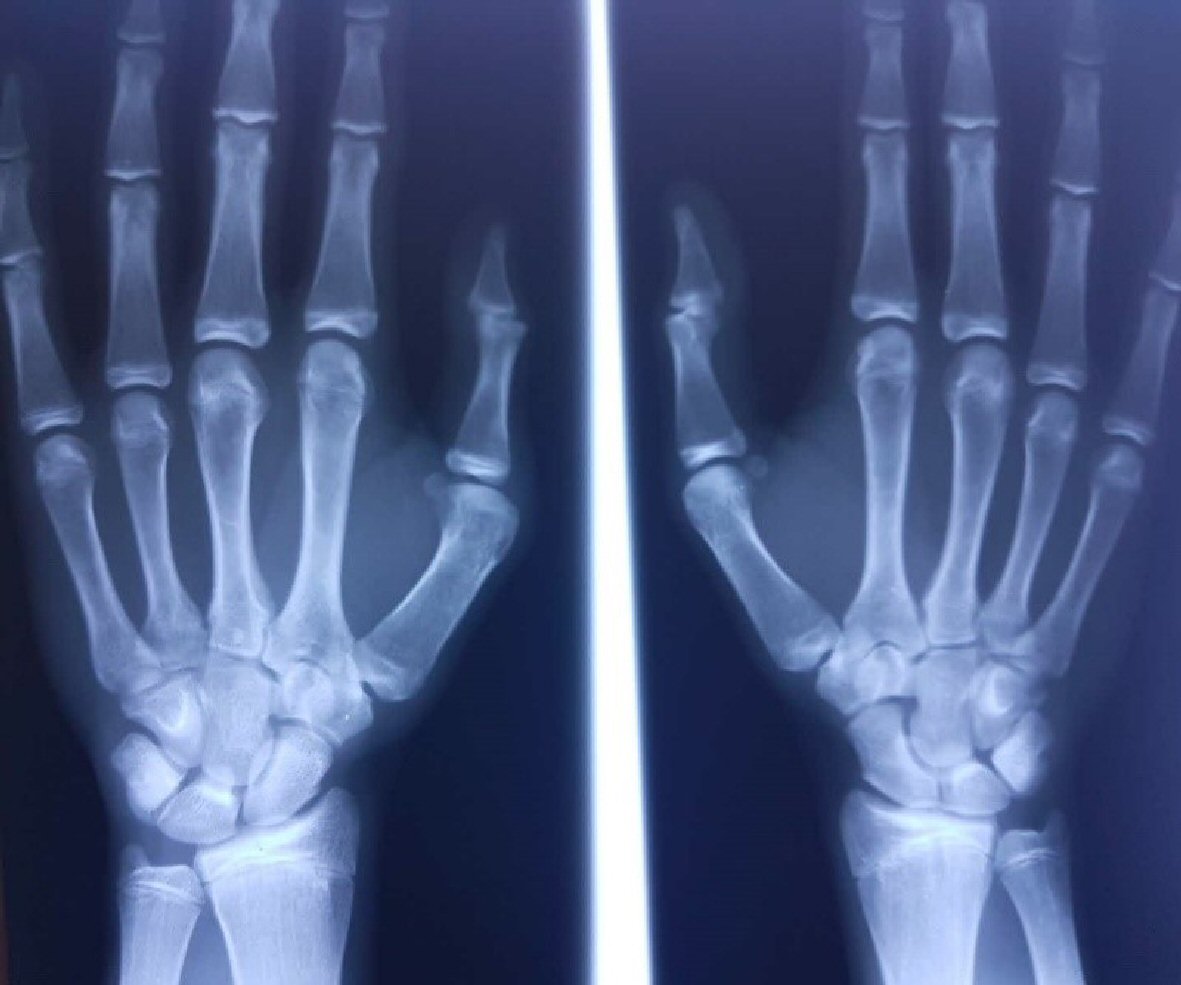 Click for large image | Figure 7. Posteroanterior hand radiograph: the index case, an 18 years and 5 months boy, with a bone age corresponding to 16 years. |
Genetic testing was done by MLPA using the SALSA MLPA P180 Limb malformations-2 probe mix (MRC-Holland, Amsterdam, Netherlands). The result was normal and duplications or deletions in the nine exons of the TBX5 gene were ruled out. The patient will be tested by sequence analysis in order to detect the pathogenic variant in TBX5.
Two of the family members (mother and sister’s proband) are also screened by clinical exam, ECG, TTE and posteroanterior radiograph of both hands. All these were without pathological changes. The father and the old brother were not examined because they are abroad, but there are no significant antecedents for HOS.
| Discussion | ▴Top |
The management of patients with HOS involves a multidisciplinary team consisting of a specialist in cardiology, orthopedics (especially specialist in hand surgery), medical genetics. Often the specialist in hand surgery is the one that comes first in contact with such a patient.
Radial defect is the most common deficiency of the upper limb (prevalence 1:30000). It is characterized by the unique or combined presence of the following signs: impaired radius (hypo- or aplasia), carpal bones (scaphoid and trapezium), thumb and metacarpal bone of the thumb [14]. In two-thirds of cases are associated with other congenital defects. Preaxial polydactyly is defined as the presence of additional fingers of the radial/tibial side of hand/foot. The incidence is 0.08 - 1.4 in 1,000 live births [15]. Manifestations are bilaterally in the vast majority of cases, and the left hand is severely impaired compared to the right one. Because ossification is incomplete in childhood, it is impossible to highlight changes in carpal bones at this time.
In our case it seems to be polydactyly type IV (Wassel classification) with complete duplication of both phalanges but with only one metacarpal bone. The abnormalities of carpal bones are bilateral, but supplementary thumb was present only on the right side. The carpal bones changes were: bilateral hypertrophic pisiform, small bilateral trapezium and trapezoid bones and delayed bone age. An abnormal large pisiform was described in some cases of HOS [16]. Delayed bone age was a radiological sign in others patients with HOS [17].
CHD are one of the most frequent congenital abnormalities in 1% to 2-3% newborns (if minor defects are included). The incidence of CHD in HOS is high (75%). The clinical effects could be severe. In complex CHD the mortality remained high, despite the progresses of cardiac surgery. More defects were identified in our patient: ASD, bicuspid aortic valve, LV non-compaction. ASD is the most common CHD in HOS. An associated finding in this patient was the LV non-compaction meeting Jenni criteria. A few cases of LV non-compaction in HOS have been reported in the literature, apparently not determined determined by TBX5 mutation, but further studies are needed [18]. The risk consists in the development of subsequent cardiomyopathy, arrhythmias and thromboembolism [19].
Patients are at risk for cardiac conductive defects, mainly sinus bradycardia, first-degree atrioventricular block or right bundle branch block. In this case, we noted intraventricular conduction disorder manifesting as slurring of QRS in aVL and V3. Atrial fibrillation, atrial flutter or sudden cardiac deaths have also been reported [16].
HOS is inherited in an autosomal dominant manner, but just in 15% of cases the patient has an affected parent. The family history is negative for HOS, but it may be apparently negative because of failure to recognize the signs and symptoms of HOS, early death of a person before the onset of symptoms, and mild phenotype with late onset of symptoms. The genetic tests are recommended to all first-degree relatives if the mutation is known in proband, otherwise the tests like ECG, TTE and wrist radiograph are recommended for the screening of clinical changes. Posteroanterior wrist radiograph is very important because the carpal bones abnormalities are present in 100% of the cases and sometimes are the unique sign of this disorder [12]. Mother and sister are normal in our cases, and father and brother will be investigated.
Genetic testing by MLPA was normal in our patient, but this result does not rule out the HOS because this technique does highlight only TBX5 gene deletions/duplications and on the other hand the pathogenic variant was identified just in 74% of cases with strict clinical criteria [20].
HOS is a pleiotropic disorder, in which the main anatomic regions implicated are the heart and the upper limbs. Because of variable expressivity the diagnosis is sometimes delayed. An early diagnosis is necessary for preventing complications in patients and for identifying other family members at risk.
Conflict of Interest
The authors have no conflict of interest to declare.
| References | ▴Top |
- Barisic I, Boban L, Greenlees R, Garne E, Wellesley D, Calzolari E, Addor MC, et al. Holt Oram syndrome: a registry-based study in Europe. Orphanet J Rare Dis. 2014;9:156.
doi pubmed - Zaragoza MV, Hakim SA, Hoang V, Elliott AM. Heart-hand syndrome IV: a second family with LMNA-related cardiomyopathy and brachydactyly. Clin Genet. 2017;91(3):499-500.
doi pubmed - Orphanet Report Series - Prevalence of rare diseases: Bibliographic data - January 2018 - Number 1. Available from: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf.
- Ghosh TK, Brook JD, Wilsdon A. T-Box genes in human development and disease. Curr Top Dev Biol. 2017;122:383-415.
doi pubmed - Postma AV, Christoffels VM, Bezzina CR. Developmental aspects of cardiac arrhythmogenesis. Cardiovasc Res. 2011;91(2):243-251.
doi pubmed - Nerbonne J. Molecular analysis of voltage-gated K+ channel diversity and functioning in the mammalian heart. In: Terjung R, ed. Comprehensive Physiology. Wiley. 2010: 568-594.
- Russell MW, Chung WK, Kaltman JR, Miller TA. Advances in the understanding of the genetic determinants of congenital heart disease and their impact on clinical outcomes. J Am Heart Assoc. 2018;7(6):e006906.
doi pubmed - Hiroi Y, Kudoh S, Monzen K, Ikeda Y, Yazaki Y, Nagai R, Komuro I. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat Genet. 2001;28(3):276-280.
doi pubmed - Waldron L, Steimle JD, Greco TM, Gomez NC, Dorr KM, Kweon J, Temple B, et al. The cardiac TBX5 interactome reveals a chromatin remodeling network essential for cardiac septation. Dev Cell. 2016;36(3):262-275.
doi pubmed - Veien ES, Rosenthal JS, Kruse-Bend RC, Chien CB, Dorsky RI. Canonical Wnt signaling is required for the maintenance of dorsal retinal identity. Development. 2008;135(24):4101-4111.
doi pubmed - Darwich R, Li W, Yamak A, Komati H, Andelfinger G, Sun K, Nemer M. KLF13 is a genetic modifier of the Holt-Oram syndrome gene TBX5. Hum Mol Genet. 2017;26(5):942-954.
doi pubmed - McDermott DA, Fong JC, Basson CT. Holt-Oram Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. GeneReviews((R)). Seattle (WA), 1993.
- Basson CT, Cowley GS, Solomon SD, Weissman B, Poznanski AK, Traill TA, Seidman JG, et al. The clinical and genetic spectrum of the Holt-Oram syndrome (heart-hand syndrome). N Engl J Med. 1994;330(13):885-891.
doi pubmed - Pakkasjarvi N, Koskimies E, Ritvanen A, Nietosvaara Y, Makitie O. Characteristics and associated anomalies in radial ray deficiencies in Finland - a population-based study. Am J Med Genet A. 2013;161A(2):261-267.
doi pubmed - Comer GC, Potter M, Ladd AL. Polydactyly of the hand. J Am Acad Orthop Surg. 2018;26(3):75-82.
doi pubmed - Poznanski AK, Gall JC, Jr., Stern AM. Skeletal manifestations of the Holt-Oram syndrome. Radiology. 1970;94(1):45-53.
doi pubmed - Joshi A, Narayan JP, Garg P, Narayan S. Two unrelated families of Holt Oram syndrome: Delayed Bone Age, PDA and Complex CHD as unreported features. Int J Sci Stud. 2015;3(2):240-244.
- Ross SB, Bagnall RD, Yeates L, Sy RW, Semsarian C. Holt-Oram syndrome in two families diagnosed with left ventricular noncompaction and conduction disease. HeartRhythm Case Rep. 2018;4(4):146-151.
doi pubmed - Petersen SE, Selvanayagam JB, Wiesmann F, Robson MD, Francis JM, Anderson RH, Watkins H, et al. Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol. 2005;46(1):101-105.
doi pubmed - McDermott DA, Bressan MC, He J, Lee JS, Aftimos S, Brueckner M, Gilbert F, et al. TBX5 genetic testing validates strict clinical criteria for Holt-Oram syndrome. Pediatr Res. 2005;58(5):981-986.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cardiology Research is published by Elmer Press Inc.